

Descodificando el genoma

El genoma es un código formado por las secuencias de cuatro nucleótidos representados por las letras G, A, C y T. Desde que se conoció, mucha gente ha estado investigando cómo descodificar el genoma. Y, por supuesto, para descodificar, primero hay que leer esas letras o, dicho de otro modo, secuenciar el genoma. Es decir, hay que saber en qué orden están esas G, A, C, y esas T.
Para ello se ha utilizado sobre todo un método en los últimos 30 años, si bien durante todos estos años ha experimentado mejoras significativas. Este método fue desarrollado por Frederick Sanger y sus compañeros en la década de los 70 --por lo que recibió el segundo Premio Nobel -.
La reacción básica del método de Sanger requiere cuatro componentes principales: la porción de ADN que se desea secuenciar o "patrón de ADN", cuatro tipos de nucleótidos libres, los pequeños filamentos de ADN de 20-30 nucleótidos conocidos como iniciadores o primeros, y la enzima de ADN polimerasa que sintetiza el ADN.
La reacción comienza calentando el patrón de ADN para que se separen los dos filamentos. De esta forma, el iniciador se unirá a uno de los filamentos (donde hay una secuencia adicional). Porque la polimerasa DNA alarga los filamentos de ADN, pero no se puede empezar por la nada, por eso es necesario el iniciador. Una vez que el iniciador se ha unido al patrón de ADN, el ADN polimerasa iniciadora comienza a estirarse nucleótido: donde hay una A en el patrón pondrá una T y viceversa, y donde hay una G una C y viceversa. Esto ocurre, además, siempre en un sentido determinado, es decir, los dos extremos de un filamento de ADN se denominan 5' y 3', y la polimerasa DNA añade nucleótidos al extremo 3'.
Por eso, si partiéramos de varias copias de un patrón de ADN, obtendríamos otras tantas copias de uno de sus filamentos. Sin embargo, la clave del método de Sanger está en el uso de algunos nucleótidos modificados: los dideoxinucleótidos. A estos nucleótidos transformados les falta un grupo de hidroxilos en el extremo 3', lo que hace que no puedan añadirse otros nucleótidos.

Cuatro nucleótidos, cuatro reacciones
El método de Sanger requiere cuatro reacciones para cada patrón de ADN. En las cuatro reacciones se encuentran los cuatro componentes mencionados anteriormente, pero en cada una de ellas se coloca un único dideoxinucleótido. Así, por ejemplo, una de las reacciones contendrá cuatro nucleótidos normales y algunos dideoxi-G. En esta reacción, cuando el ADN polimerasa iniciador comienza a estirarse, cada vez que necesita un G, pueden ocurrir dos cosas aleatorias: Tomar una G normal o una dideoxi-G. La mayoría son normales G, pero cuando toma un dideoxi-G, la polimerasa no podrá seguir estirando la cadena. Así, a partir de varias copias del patrón, obtendremos filamentos de diferente longitud, terminados todos en un G. Y a partir de millones de copias, finalmente tendremos los filamentos correspondientes a cada G de la secuencia. Y lo mismo ocurre con las otras tres reacciones.
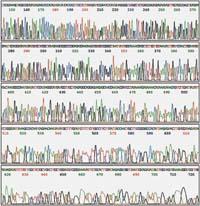
Posteriormente, los nuevos filamentos obtenidos se separan por tamaños mediante una electroforesis. En la electroforesis, los filamentos de ADN se colocan en un campo eléctrico y como el ADN tiene carga negativa se desplazan del polo negativo al positivo. En el camino se coloca algo que dificulte este movimiento, como un gel poroso, de manera que las partes grandes tardan más tiempo en moverse de un polo a otro que las pequeñas. De esta forma, se pueden distinguir los filamentos que tienen un lado de un nucleótido.
Para poder visualizar el resultado de la electroforesis, los nuevos filamentos deben estar de alguna manera marcados. Inicialmente se utilizaban nucleótidos o iniciadores marcados radiactivamente o fluorescentes. Así, mediante una autoradiografía, o mediante rayos ultravioleta, se pueden ver como bandas, filamentos separados por tamaños.
Cada reacción consiste en trazar una línea en la electroforesis, y el resultado final es que veremos una banda por cada nucleótido de la secuencia en una de las cuatro líneas, y así, siguiendo el orden de dichas bandas, podremos leer dicha secuencia.

Automatización
Esta es básicamente la técnica utilizada para secuenciar la mayor parte del ADN secuenciado hasta el momento. Pero desde su fundación, Sanger ha experimentado importantes mejoras. Uno de los más importantes fue la secuenciadora automática de ADN inventada por Leroy Hood en 1986. Hood marcó cada uno de los cuatro dideoxinucleótidos de forma que proporcionara una fluorescencia de diferente longitud de onda (color). Así, por un lado, en lugar de cuatro reacciones, basta con una sola, y por otro, la longitud de onda emitida al ser iluminada con rayos ultravioleta se puede detectar automáticamente.
En los secuenciadores automáticos, cada muestra va en una sola línea, detectándose los cuatro nucleótidos por su diferente color. En los secuenciadores modernos, los filamentos de ADN se separan pasando por unos capilares de fibra de vidrio, a la salida del extremo son golpeados por un láser y la máquina detecta la presencia de fluoerescencia. Los filamentos que salen primero serán el iniciador más un nucleótido y a continuación saldrá toda la secuencia.
Durante la última década, los secuenciadores automáticos de ADN han mejorado considerablemente, pudiendo procesar muchas más muestras con mayor rapidez y facilitando su uso. En la actualidad se pueden secuenciar cientos de muestras en una sesión y en un día se pueden realizar unas 24 sesiones.
Genoma grande

Sin embargo, todavía no es tarea fácil descodificar todo un genoma. Las técnicas de secuenciación del ADN sirven para secuenciar pequeños fragmentos de ADN, secuencias de 300-900 nucleótidos. De hecho, en filamentos más largos, no pueden separarse con precisión de la cara de un nucleótido.
Pero el genoma pequeño de una bacteria también contiene millones de nucleótidos, y el humano 3.000.000.000, dividido en 23 cromosomas. Imagínate que para escribir todo el genoma letra a letra en esta revista necesitaríamos unas 20.000 revistas. La longitud media de las secuencias leídas (500 nucleótidos) sería el siguiente párrafo, y en 20.000 revistas se incluyen seis millones de párrafos de este tipo. Además, para conseguir una fiabilidad suficiente, una vez no, habría que leer entre 6 y 10 veces todas estas revistas.
Es lo que hicieron en el Proyecto de Genoma Humano. Por partes leyeron todo el genoma humano letra a letra. Sin embargo, dividir todo el genoma y leer todas estas partes es una cosa y la otra es saber el orden en el que se deben unir esas partes.
El proyecto comenzó con la construcción de un mapa del genoma. Para poder recorrer los cromosomas sin perder, se tomaron miles de puntos de referencia. Una vez realizados estos mapas, se creó una "biblioteca" de fragmentos de ADN que englobaría todo el genoma. Para ello, estos trozos de ADN fueron almacenados en el interior de las bacterias, y gracias a los puntos de referencia, cada parte del genoma era conocida. En definitiva, es lo que ofrece cualquier biblioteca: información ordenada. Esto ha permitido trabajar de forma coordinada en laboratorios de todo el mundo.
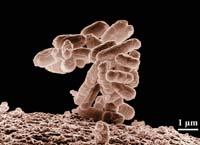
Los fragmentos de ADN fueron almacenados en las bacterias E. coli que viven habitualmente en nuestros intestinos, como cromosomas bacterianos artificiales (BAC). En cada BAC se almacenan fragmentos de ADN de 100.000 o 200.000 nucleótidos.
Las bacterias E. coli conservan el tiempo que quieras en el congelador. Así, cuando el científico necesita un BAC de la biblioteca, la bacteria sólo tiene que resucitar a 37ºC. Además, cuando la bacteria que contiene el fragmento de ADN se reproduce con él. Así, basta con que las bacterias dejen crecer una noche para obtener millones de copias de su parte interna de ADN. A esto se le llama amplificación del ADN.
Los BACs son todavía muy grandes para poder secuenciar. Por ello, los BACs también se dividen aleatoriamente, obteniendo partes más pequeñas que se solapan. Estas partes se introducen en los virus o plásmidos que infectan las bacterias para su posterior amplificación en las bacterias E. coli. Por último, el ADN de las bacterias es purificado y secuenciado. A continuación se identifican las secuencias que se solapan en las distintas partes y se completa la secuencia completa del BAC. Al mismo tiempo, los BACs también se superponen. Y así, la lectura por lectura, el BAC por BAC y el cromosoma por cromosoma, hasta leer todo el genoma.
Carrera genómica
Leer así un genoma completo requiere mucho tiempo, trabajo y dinero. Por ello, los investigadores están investigando continuamente para desarrollar técnicas de secuenciación más rápidas y baratas. Muchos nuevos métodos buscan aumentar la capacidad de secuenciación y realizar miles o millones de secuencias simultáneamente. Pero tampoco faltan ideas nuevas, como un método que detecta en el momento en que la dna-polimerasa añade el nucleótido al filamento, o que detecta corrientes eléctricas que identificarían cada nucleótido al pasar los filamentos por nanopors.
Los proyectos son muchos y se están desarrollando a una velocidad vertiginosa. No es de extrañar que las instituciones públicas y privadas estén invirtiendo mucho. En 2006, el National Human Genome Research Institute (NHGRI) de EE.UU. aportó 13 millones de dólares a la financiación de proyectos para acelerar el desarrollo de tecnologías que abaraten la secuenciación de ADN. Ese mismo año, la Fundación X Prize anunció un premio de diez millones de dólares para el primer equipo que en 2006 realiza un dispositivo capaz de secuenciar cien genomas humanos en diez días. Y, cien no, un proyecto internacional que acaba de lanzar pretende secuenciar los genomas de mil seres humanos.
Se ha dicho que la secuenciación del genoma es uno de los avances más importantes en la historia del ser humano. Investigadores de todo el mundo intentan desvelar todos los secretos del genoma y descodificar el código. Está claro que estamos en la era de la genómica.

Zu idazle
Zientzia aldizkaria

