

Decoding the genome

The genome is a code formed by the sequences of four nucleotides represented by the letters G, A, C and T. Since it became known, many people have been investigating how to decode the genome. And, of course, to decode, you must first read those letters or, in other words, sequence the genome. That is, we must know in what order are those G, A, C, and those T.
For this purpose, a method has been used mainly in the last 30 years, although during all these years it has experienced significant improvements. This method was developed by Frederick Sanger and his companions in the 1970s -- so he received the second Nobel Prize.
The basic reaction of the Sanger method requires four main components: the portion of DNA that is wanted to sequence or "DNA pattern", four types of free nucleotides, the small 20-30 nucleotides DNA filaments known as initiators or first, and the polymerase DNA enzyme that synthesizes DNA.
The reaction begins by heating the DNA pattern to separate the two filaments. In this way, the initiator will join one of the filaments (where there is an additional sequence). Because polymerase DNA lengthens the filaments of DNA, but you can not start at all, that is why the initiator is necessary. Once the initiator has joined the DNA pattern, the initiating polymerase DNA begins to stretch nucleotide: where there is an A in the pattern will put a T and vice versa, and where there is a G a C and vice versa. This also occurs, always in a certain sense, that is, the two ends of a DNA filament are called 5' and 3', and the polymerase DNA adds nucleotides to the end 3'.
Therefore, if we split from several copies of a pattern of DNA, we would get as many copies of one of its filaments. However, the key to the Sanger method is in the use of some modified nucleotides: dideoxynucleotides. These transformed nucleotides lack a group of hydroxyl at the end 3', which means that other nucleotides cannot be added.

Four nucleotides, four reactions
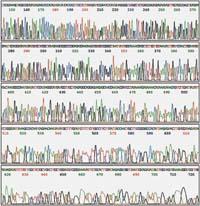
The Sanger method requires four reactions for each DNA pattern. In the four reactions are the four components mentioned above, but in each of them a single dideoxynucleotide is placed. Thus, for example, one of the reactions will contain four normal nucleotides and some dideoxi-G. In this reaction, when the initiating polymerase DNA begins to stretch, every time you need a G, two random things can occur: Take a normal G or a dideoxi-G. Most are normal G, but when you take a dideoxi-G, polymerase will not be able to continue stretching the chain. Thus, from several copies of the pattern, we will obtain filaments of different length, all finished in a G. And from millions of copies, we will finally have the filaments corresponding to each G of the sequence. And the same happens with the other three reactions.
Subsequently, the new filaments obtained are separated by sizes by an electrophoresis. In electrophoresis, DNA filaments are placed in an electrical field and as DNA has negative charge they move from the negative pole to the positive. On the way is placed something that hinders this movement, like a porous gel, so that large parts take longer to move from one pole to another than small ones. In this way, the filaments that have a side of a nucleotide can be distinguished.
In order to visualize the result of the electrophoresis, the new filaments must be in some way marked. Initially nucleotides or initiators marked radiactively or fluorescent. Thus, by an autoradiography, or by ultraviolet rays, they can be seen as bands, filaments separated by sizes.
Each reaction consists of tracing a line in the electrophoresis, and the final result is that we will see a band for each nucleotide of the sequence in one of the four lines, and thus, following the order of these bands, we can read this sequence.

Automation Automation Automation Automation
This is basically the technique used to sequence most of the sequenced DNA so far. But since its foundation, Sanger has experienced significant improvements. One of the most important was the automatic DNA sequencer invented by Leroy Hood in 1986. Hood marked each of the four dideoxynucleotides in a way that provided a fluorescence of different wavelength (color). Thus, on the one hand, instead of four reactions, just one, and on the other, the wavelength emitted when illuminated with ultraviolet rays can be detected automatically.
In the automatic sequencers, each sample goes on a single line, detecting the four nucleotides by their different color. In modern sequencers, the DNA filaments are separated by a glass fibre capillaries, at the exit of the end they are hit by a laser and the machine detects the presence of fluoerescence. The filaments that come out first will be the initiator plus a nucleotide and then the whole sequence will come out.
During the last decade, automatic DNA sequencers have improved considerably, being able to process many more samples faster and facilitating their use. Currently, hundreds of samples can be sequenced in one session and in one day, about 24 sessions can be carried out.
Large genome

However, it is not yet easy task to decode an entire genome. The DNA sequencing techniques serve to sequence small fragments of DNA, sequences of 300-900 nucleotides. In fact, in longer filaments, they cannot be precisely separated from the face of a nucleotide.
But the small genome of a bacterium also contains millions of nucleotides, and the human 3,000,000,000, divided into 23 chromosomes. Imagine that to write all the genome lyrics in this magazine we would need about 20,000 magazines. The average length of read sequences (500 nucleotides) would be the following paragraph, and 20,000 journals include six million paragraphs of this type. In addition, to achieve sufficient reliability, once not, we would have to read between 6 and 10 times all these magazines.
This is what they did in the Human Genome Project. In parts they read the entire human genome letter by letter. However, dividing the entire genome and reading all these parts is one thing and the other is to know the order in which those parts should be joined.
The project began with the construction of a genome map. In order to travel the chromosomes without losing, thousands of reference points were taken. Once these maps were made, a "library" of DNA fragments was created that would encompass the entire genome. To do this, these pieces of DNA were stored inside the bacteria, and thanks to the reference points, each part of the genome was known. In short, this is what any library offers: orderly information. This has allowed to work in a coordinated way in laboratories around the world.
Fragments of DNA were stored in E. coli bacteria that usually live in our intestines, such as artificial bacterial chromosomes (BAC). In each BAC fragments of DNA of 100,000 or 200,000 nucleotides are stored.
The E. coli bacteria keep as long as you want in the freezer. Thus, when the scientist needs a BAC from the library, the bacterium only has to resurrect at 37ºC. In addition, when the bacterium containing the fragment of DNA reproduces with it. So, it's enough for bacteria to grow one night to get millions of copies of their inner part of DNA. This is called DNA amplification.
The BACs are still very large for sequencing. Therefore, BACs are also divided randomly, obtaining smaller parts that overlap. These parts are introduced into viruses or plasmids that infect bacteria for further amplification in E. coli bacteria. Finally, the DNA of bacteria is purified and sequenced. The sequences that overlap in the different parts are identified below and the complete sequence of the BAC is completed. At the same time, BACs also overlap. And so, reading by reading, BAC by BAC and chromosome by chromosome, until reading the entire genome.
Genomic career
Reading this way a complete genome requires a lot of time, work and money. For this reason, researchers are continually investigating to develop faster, cheaper sequencing techniques. Many new methods seek to increase sequencing capacity and perform thousands or millions of sequences simultaneously. But there is also no shortage of new ideas, such as a method that detects when the dna-polymerase adds the nucleotide to the filament, or detects electrical currents that would identify each nucleotide by passing the filaments by nanopors.
The projects are many and are being developed at a dizzying speed. It is no wonder that public and private institutions are investing a lot. In 2006, the US National Human Genome Research Institute (NHGRI). contributed $13 million to project finance to accelerate the development of technologies that cover the sequencing of DNA. That same year, the X Prize Foundation announced a prize of ten million dollars for the first team that in 2006 makes a device capable of sequencing one hundred human genomes in ten days. And, one hundred no, an international project that just launched aims to sequence the genomes of a thousand human beings.
It has been said that genome sequencing is one of the most important advances in human history. Researchers from all over the world try to reveal all the secrets of the genome and decode the code. It is clear that we are in the age of genomics.

Zu idazle
Zientzia aldizkaria

