

Reciclar proteínas, chave paira estar san
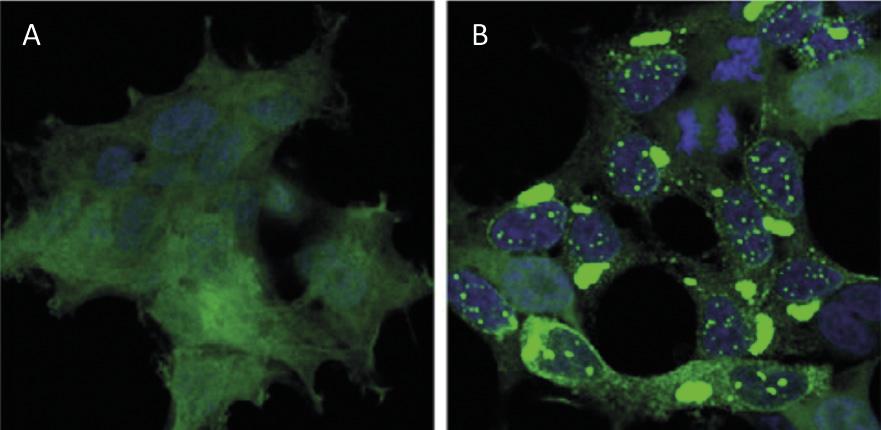
A reciclaxe de proteínas a nivel celular ten un impacto similar ao da reciclaxe de residuos a nivel mundial. As proteínas son as principais moléculas que garanten o funcionamento das células. Son responsables de levar a cabo reacciones químicas, regular os xenes, transportar moléculas e manter a estrutura celular. Cada célula contén miles de proteínas que ademais se forman de forma moi dinámica. De feito, paira responder as necesidades da célula actual ou aos estímulos externos, está a producirse e destruíndo constantemente proteínas de diferentes tipos celulares. Por tanto, é imprescindible manter o equilibrio entre estes dous procesos adversos paira garantir o correcto funcionamento da célula. Por iso, non é de estrañar que moitos dos procesos de sínteses e degradación de proteínas que azoutan a nosa sociedade estean na orixe de moitas enfermidades. Entre as enfermidades desenvolvidas como o Alzheimer, o Parkinson e o Huntington, todas elas moi coñecidas [1,2] (Figura 1).
Proteínas: ben encartadas ou acabadas.
A información paira a produción de proteínas está almacenada nos xenes. Na cadea de ADN especifícase cando e que proteína xerarase. A estrutura dunha proteína que se extrae da fábrica de produción de proteínas, é dicir, dos ribosomas, é equivalente a un colar de perlas con aminoácidos. Con todo, esta proteína, paira ser funcional, debe ter una determinada estrutura tridimensional. Na célula pódense atopar 20 tipos de aminoácidos e a secuencia de aminoácidos de cada proteína determina a estrutura funcional tridimensional, é dicir, a estrutura nativa desta proteína. Os requisitos celulares son moi especiais, polo que as proteínas necesitan a miúdo a axuda de proteínas denominadas chaperones moleculares paira obter una estrutura nativa. Os seres humanos temos máis de cen chapperones, cada un dos cales se encarga do proceso de encartado de centos de proteínas ituales. Cando a actividade dos txaperones está danada polas mutacións, o proceso de encartado das proteínas diana non é o adecuado, polo que estas proteínas non son funcionais. Se non se degradan, acumúlanse na célula causando graves danos [4] (Figura 2).
Por exemplo, as mutacións ocorridas no txaperón denominado HSPB1 provocan una esclerose lateral amiotrófica e una neuropatía grave denominada Charcot Marie Tooth [5,6]. E os que teñen o chaperón CHIP mutado desenvolven a síndrome de Gordon Holmes, un mal único que causa hipogonadismo e problemas neurológicos.
Pero en todos os casos nos que as proteínas se dobran mal e acumúlanse patológicamente, a culpa non é dos txaperos. Nalgúns casos, debido ás mutacións que se producen no xenoma, as proteínas prodúcense con máis aminoácidos dos necesarios, o que impide obter a estrutura nativa correspondente (Figura 2).
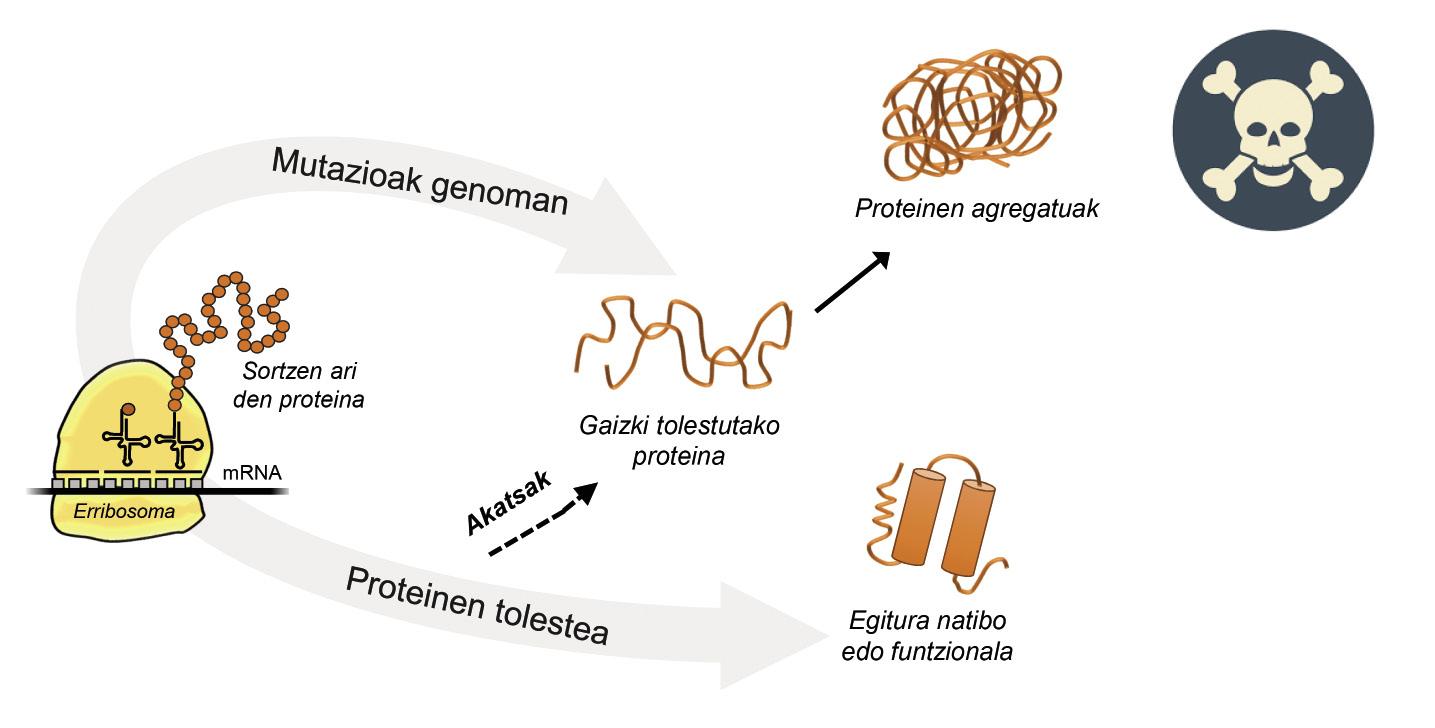
Por exemplo, os afectados pola enfermidade de huntington teñen una proteína de huntingtina defectuosa. De feito, ao mutarse o xene HTT que codifica a huntingtina, a proteína producida contén máis aminoácidos glutamina dos necesarios, é dicir, contén demasiados exemplares dunha perla determinada [4]. Os afectados pola esclerose lateral múltiple e a demencia frontotemporal tamén presentan una proteína C9orf72 defectuosa con máis pares de aminoácidos glicina dos necesarios, que ten una gran tendencia a acumularse en neuronas [7].
Con todo, a enfermidade máis coñecida pola acumulación de proteínas é o alzheimer, una enfermidade que axita fortemente a sociedade actual. A medida que morren as neuronas do hipocampo cerebral, as persoas enfermas perden as súas capacidades cognitivas, especialmente a memoria a curto prazo. Pero, por que morren as neuronas? Cada vez hai máis evidencia de que as acumulacións de proteínas tau e beta-amiloide mutadas son as principais responsables da morte das neuronas [6].
Proteasoma: trituradora de proteínas
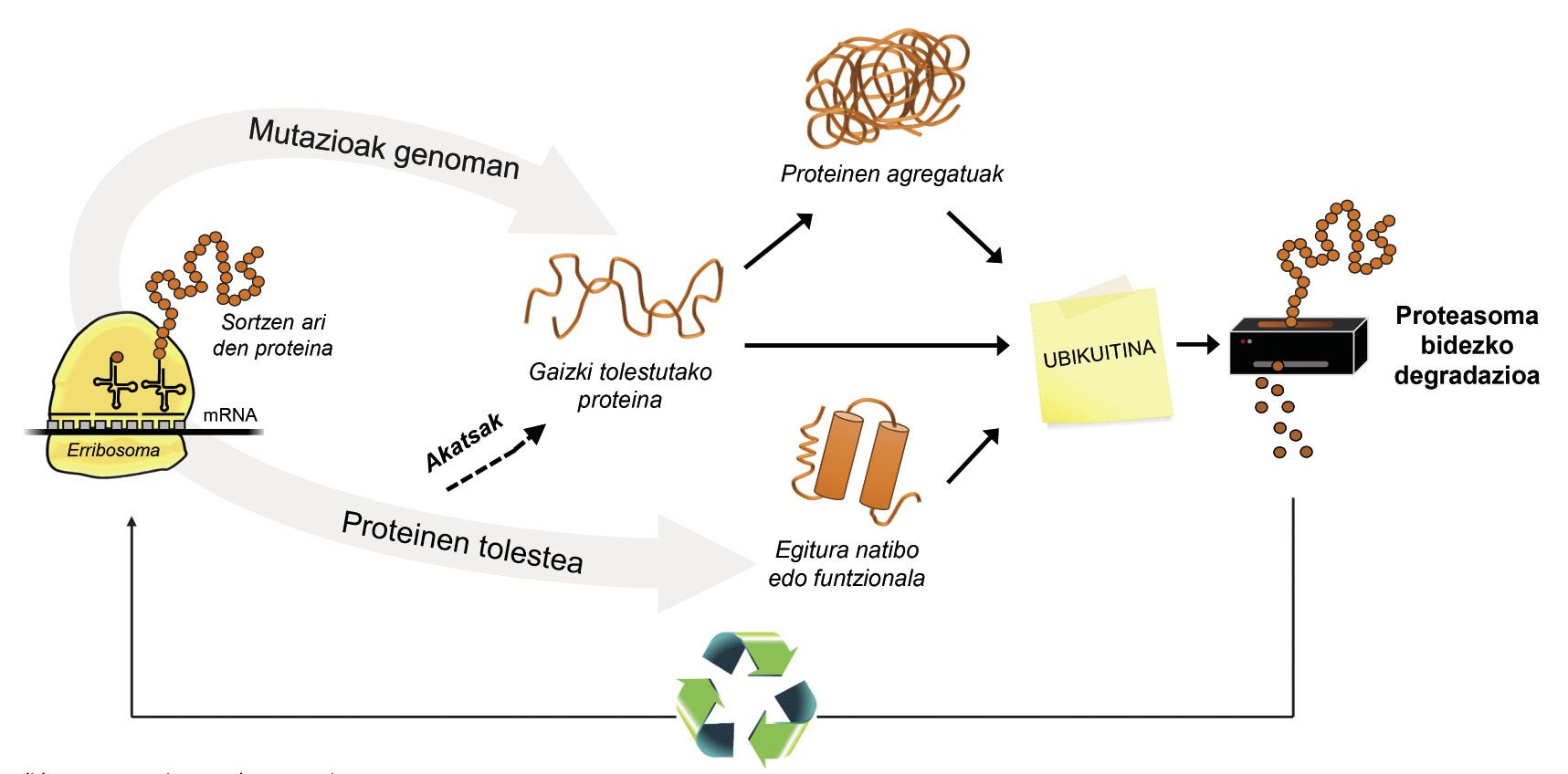
E a célula non ten mecanismos paira degradar proteínas mal encartadas e evitar agregados patolóxicos? Si, e máis dunha. Con todo, por unha banda, tendo en conta os exemplos anteriormente mencionados, é evidente que algunhas acumulacións de proteínas son capaces de romper con estes mecanismos, e doutra banda, a propia máquina de degradación de proteínas pode ter problemas e impedir a reciclaxe.
Ademais das proteínas mal encartadas, a maioría das proteínas non dispoñibles paira a célula degrádanse grazas a un complexo multiproteico chamado proteasoma. Pero entre os miles de proteínas que hai na célula, como se pode saber cal é o que hai que degradar? Como percibe o proteasoma que proteínas hai que reducir? Pois porque teñen adherida una pequena molécula chamada ubiquitina. Esta molécula funciona como un post-it que di “debes ir a proteasoma a degradar”. Proteasoma actúa como una trituradora de papel. Reduce as proteínas, é dicir, rompe as relacións entre os aminoácidos. Paralelamente, pon a disposición da célula aminoácidos libres paira a produción de novas moléculas [8]. Por iso, pódese afirmar que o proteasoma recicla proteínas (Figura 3).
O proceso de reciclaxe de proteínas é moi complexo e abonda con que se produza un erro en calquera punto para que se desenvolva una enfermidade. No proceso de unión da ubiquitina ás proteínas que van ser transferidas a proteasoma, colaboran tres tipos de encimas: Encimas E1, E2 e E3. Si por estar mutadas estas encimas o proceso de ubiquitinación das proteínas non é correcto, as proteínas a degradar non se destrúen e acumúlanse na célula. Esta toxicidade maniféstase sobre todo en neuronas altamente vulnerables, o que significa que moitas enfermidades neurológicas están relacionadas cos defectos que se producen nestas encimas [8].
A encima UBA1 tipo E1, por exemplo, está relacionada cunha enfermidade neuromuscular excepcional que só afecta os mozos. Estes pacientes, ademais de ter una gran debilidade muscular, non teñen reflexos, é dicir, os músculos non responden adecuadamente a estímulos externos. Os que teñen mutada a encima UBE2A tipo E2 desenvolven a síndrome de deficiencia UBE2A, una enfermidade neurológica atípica. Esta enfermidade é tamén padecida unicamente por homes, sendo os síntomas máis frecuentes a deficiencia mental, alteracións cutáneas, do aparello reprodutor e urinario e a ausencia de fala.
Entre as tres encimas que realizan a ubiquitinación de proteínas, as máis abundantes son as E3. Así, existe algunha encima E3 mutada detrás da maioría das enfermidades neurológicas desenvolvidas como consecuencia dunha ubiquitinación defectuosa. Por exemplo, quen sofren o autismo familiar e o autismo esporádico, clasificados dentro dos trastornos do espectro do autismo, teñen mutadas as encimas UBE3B e UBE3C tipo E3, respectivamente. Pola contra, as persoas con UBE3A defectuosa desenvolven a síndrome de Angelman relacionado co autismo [9].
Ademais das enfermidades relacionadas co autismo, os E3 mutados poden causar outras enfermidades neurológicas moi diferentes. Por exemplo, quen teñen mutada a encima E3 chamada gigaxonina desenvolven a neuropatía dos axones xigantes. En condicións saudables, a gigaxonina estabiliza os neurofilamentos dos brazos neuronais, é dicir, dos axones, condicionando a súa estrutura e tamaño. Con todo, cando a gigaxonina mútase, convértese en non funcional e non vascuitina os neurofilamentos dos axones. Isto fai que non se degraden os neurofilamentos e que os axones das neuronas sexan máis grandes e longos do necesario. En consecuencia, a maioría dos pacientes que sofren esta neuropatía teñen problemas paira camiñar e, en xeral, paira coordinar os movementos [9].
Tras a recente síndrome de Tenorio atópase una encima E3, neste caso denominada RNF125. Os afectados pola síndrome de Tenorio teñen discapacidade intelectual. Ademais, este tipo de pacientes caracterízanse por un crecemento excesivo, especialmente o desenvolvemento excesivo do cranio. O contrario ocórrelles aos que teñen mutado o TRIM36, aos que lles falta gran parte do cranio e do cerebro [9].

Conclusións
É evidente que un desequilibrio no ciclo de síntese e degradación de proteínas pode producir efectos realmente nocivos paira as células e os organismos vivos en xeral. Por unha banda, proteínas mal dobradas por mutacións, acumuladas como bolsas de lixo inutilizables, poden causar a morte das células. Por outra banda, a deterioración dos dispositivos moleculares responsables da degradación e a reciclaxe de proteínas produce acumulacións tóxicas de proteínas. De forma análoga á acumulación de lixo no caso de que a actividade dunha instalación de reciclaxe de residuos anulásese.
Sabemos que uno dos principais retos da sociedade actual é a correcta xestión dos residuos. E é que, se non se garante a reciclaxe dos residuos, en poucos anos cubriremos os nosos océanos e campos con lixo e a vida farase insustentable. No mundo microscópico as células teñen un reto similar cando teñen problemas paira xestionar proteínas non funcionais. Paira garantir a súa saúde é imprescindible eliminar as proteínas tóxicas.
Por tanto, se a próxima vez esquéceseche reciclar os residuos do teu fogar, lembra o desastre que iso suporía paira una célula. De feito, a reciclaxe do lixo que xeramos os seres humanos e a reciclaxe das proteínas que se acumulan nas células son accións similares, a gran e pequena escala. Una chave para que todos poidamos manternos sans.
Bibliografía
Zu idazle
Zientzia aldizkaria

- Babesleak
-

-

Elhuyar
Nor gara | Kontaktua |
Publizitatea
| Laguntza
Pribatutasun politika | Cookien politika
ISSN-2603-6614 Elhuyar