

Recycling proteins, key to being healthy
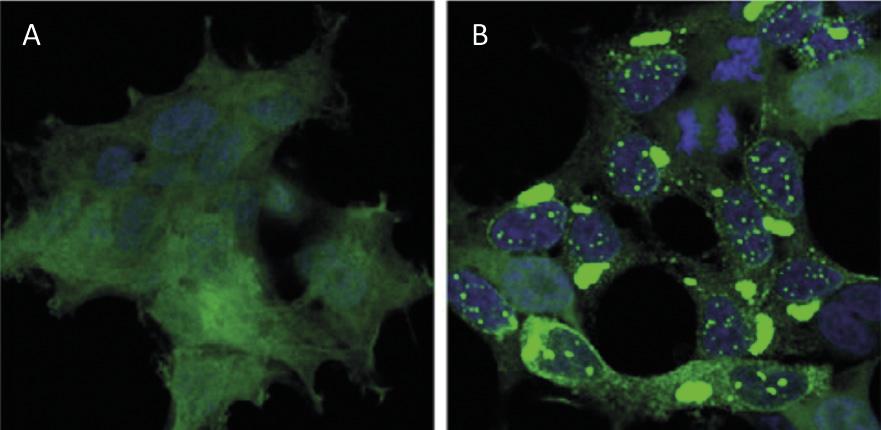
Protein recycling at the cellular level has a similar impact to waste recycling worldwide. Proteins are the main molecules that guarantee the functioning of cells. They are responsible for carrying out chemical reactions, regulating genes, transporting molecules and maintaining cell structure. Each cell contains thousands of proteins that also form very dynamically. In fact, to respond to the needs of the current cell or external stimuli, proteins of different cell types are constantly being produced and destroyed. Therefore, it is essential to maintain the balance between these two adverse processes to ensure the proper functioning of the cell. Therefore, it is not surprising that many of the processes of protein synthesis and degradation that plague our society are at the origin of many diseases. Among the diseases developed such as Alzheimer, Parkinson and Huntington, all of them well known [1,2] (Figure 1).
Proteins: either folded or finished.
Information for protein production is stored in genes. The DNA chain specifies when and what protein will be generated. The structure of a protein that is extracted from the protein production factory, that is, from ribosomes, is equivalent to a pearl necklace with amino acids. However, this protein, to be functional, must have a certain three-dimensional structure. In the cell you can find 20 types of amino acids and the amino acid sequence of each protein determines the three-dimensional functional structure, that is, the native structure of this protein. Cellular requirements are very special, so proteins often need the help of proteins called molecular chaperons to obtain a native structure. Humans have more than one hundred chapperons, each of which is in charge of the folding process of hundreds of itual proteins. When the activity of txaperones is damaged by mutations, the folding process of target proteins is not appropriate, so these proteins are not functional. If they do not degrade, they accumulate in the cell causing serious damage [4] (Figure 2).
For example, mutations in the txaperon called HSPB1 cause amyotrophic lateral sclerosis and severe neuropathy called Charcot Marie Tooth [5,6]. And those with mutated CHIP chaperon develop Gordon Holmes syndrome, a unique evil that causes hypogonadism and neurological problems.
But in all cases where proteins bend badly and accumulate pathologically, the fault is not of the txaperos. In some cases, due to mutations that occur in the genome, proteins are produced with more amino acids than necessary, which prevents obtaining the corresponding native structure (Figure 2).
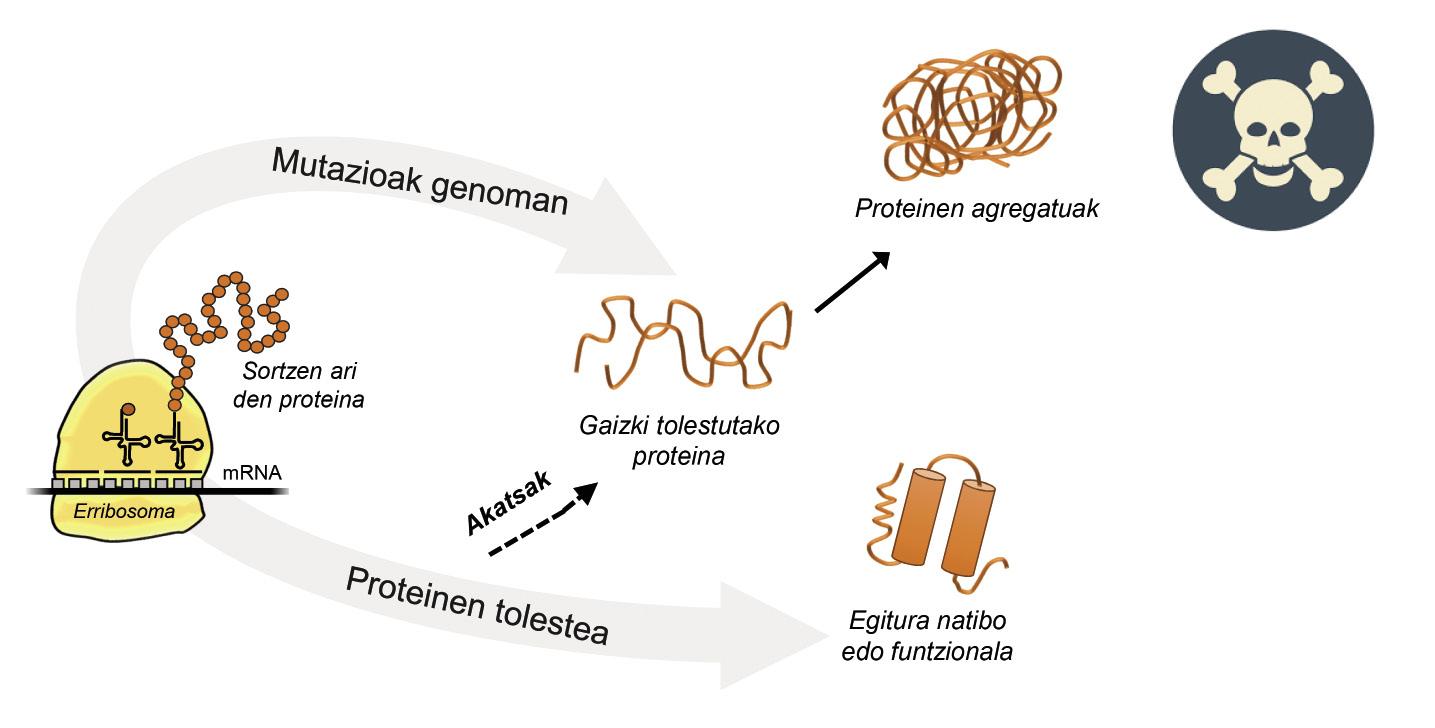
For example, those affected by huntington disease have a defective huntingtin protein. In fact, when the HTT gene encoding huntingtin is mutated, the protein produced contains more glutamine amino acids than necessary, that is, it contains too many specimens of a particular pearl [4]. Those affected by multiple lateral sclerosis and frontotemporal dementia also present a defective C9orf72 protein with more pairs of glycine amino acids than necessary, which has a great tendency to accumulate in neurons [7].
However, the disease best known for protein accumulation is Alzheimer's, a disease that strongly agitates today's society. As brain hippocampus neurons die, sick people lose their cognitive abilities, especially short-term memory. But why do neurons die? There is increasing evidence that the accumulations of mutated tau and beta-amyloid proteins are responsible for the death of neurons [6].
Proteasome: protein crusher
And does the cell have no mechanisms to degrade poorly folded proteins and avoid pathological aggregates? Yes, and more than one. However, on the one hand, taking into account the above examples, it is evident that some accumulations of proteins are able to break with these mechanisms, and on the other hand, the own protein degradation machine can have problems and prevent recycling.
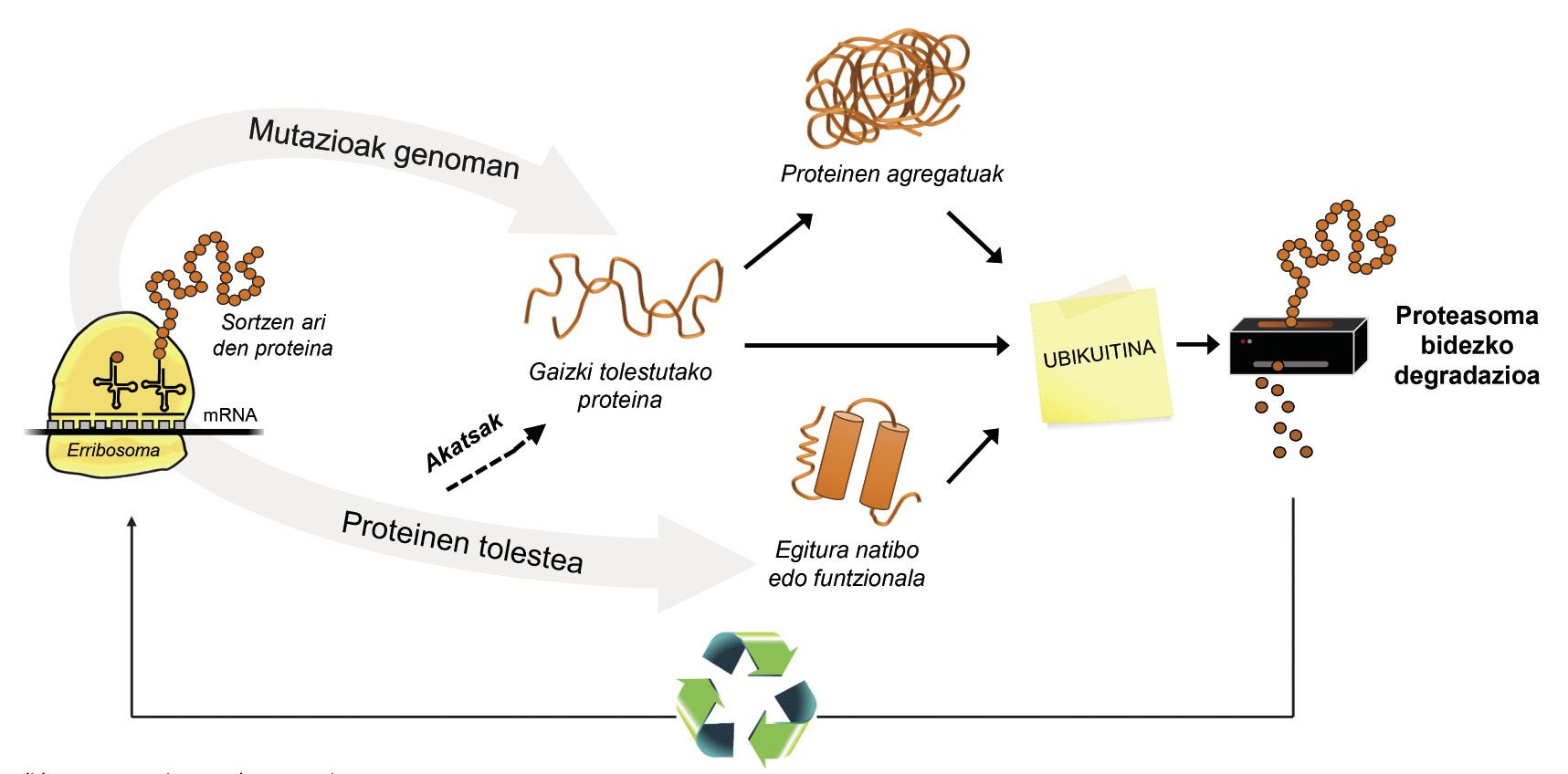
In addition to poorly folded proteins, most proteins not available to the cell are degraded by a multiprotein complex called proteasome. But among the thousands of proteins in the cell, how do you know what to degrade? How does proteasome perceive which proteins to reduce? Because they have attached a small molecule called ubiquitin. This molecule works as a post-it that says “you must go to proteasome to degrade”. Proteasoma acts as a paper crusher. It reduces proteins, that is, breaks the relationships between amino acids. At the same time, it makes available to the cell free amino acids for the production of new molecules [8]. Therefore, it can be said that proteasome recycles proteins (Figure 3).
The process of protein recycling is very complex and it is enough that an error occurs at any point for a disease to develop. In the process of binding ubiquitin to proteins to be transferred to proteasome, three types of enzymes collaborate: Enzymes E1, E2 and E3. If by being mutated these enzymes the process of ubiquitination of proteins is not correct, the proteins to degrade are not destroyed and accumulate in the cell. This toxicity is manifested mainly in highly vulnerable neurons, which means that many neurological diseases are related to the defects that occur in these enzymes [8].
The enzyme UBA1 type E1, for example, is related to an exceptional neuromuscular disease that only affects guys. These patients, in addition to having a great muscle weakness, have no reflexes, that is, the muscles do not respond adequately to external stimuli. Those who have mutated the UBE2A type E2 enzyme develop UBE2A deficiency syndrome, an atypical neurological disease. This disease is also suffered only by men, with the most frequent symptoms being mental deficiency, skin alterations, the reproductive and urinary system and the absence of speech.
Among the three enzymes that perform protein ubiquitination, the most abundant are E3. Thus, there is some mutated E3 enzyme behind most neurological diseases developed as a result of defective ubiquitination. For example, those who suffer from family autism and sporadic autism, classified within autism spectrum disorders, have mutated the enzymes UBE3B and UBE3C type E3, respectively. In contrast, people with defective UBE3A develop autism-related Angelman syndrome [9].
In addition to diseases related to autism, mutated E3 can cause other very different neurological diseases. For example, those who have mutated the enzyme E3 called gigaxonine develop the neuropathy of giant axons. In healthy conditions, gigaxonine stabilizes the neurofilaments of the neuronal arms, that is, of the axons, conditioning their structure and size. However, when gigaxonin is mutated, the neurofilaments of the axons become non-functional and non-vascuitine. This makes the neurofilaments not degrade and the axons of neurons larger and longer than necessary. Consequently, most patients suffering from this neuropathy have problems walking and, in general, coordinating movements [9].
After the recent Tenorio syndrome, an enzyme E3 is found, in this case called RNF125. Those affected by Tenorio syndrome have intellectual disabilities. In addition, these patients are characterized by excessive growth, especially the excessive development of the skull. The opposite happens to those who have mutated TRIM36, who lack much of the skull and brain [9].

Conclusions
It is evident that an imbalance in the cycle of protein synthesis and degradation can produce really harmful effects for cells and living organisms in general. On the one hand, mutation-bent proteins, accumulated as unusable garbage bags, can cause cell death. Moreover, the deterioration of molecular devices responsible for the degradation and recycling of proteins produces toxic accumulations of proteins. Similar to the accumulation of waste in case the activity of a waste recycling facility was cancelled.
We know that one of the main challenges of today's society is the correct management of waste. And if waste recycling is not guaranteed, in a few years we will cover our oceans and fields with trash and life will become unsustainable. In the microscopic world cells have a similar challenge when they have problems managing non-functional proteins. To ensure your health it is essential to eliminate toxic proteins.
So, if you forget to recycle your household waste next time, remember the disaster it would mean for a cell. In fact, the recycling of waste that humans generate and the recycling of proteins that accumulate in cells are similar actions, on a large and small scale. A key so that we can all stay healthy.
Bibliography
Zu idazle
Zientzia aldizkaria

- Babesleak
-

-

Elhuyar
Nor gara | Kontaktua |
Publizitatea
| Laguntza
Pribatutasun politika | Cookien politika
ISSN-2603-6614 Elhuyar