

Recycler les protéines, clé pour être en bonne santé
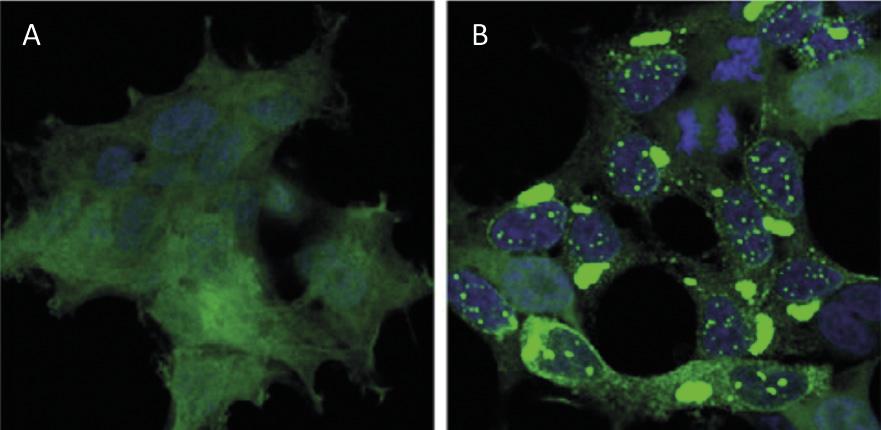
Le recyclage des protéines au niveau cellulaire a un impact similaire à celui du recyclage des déchets au niveau mondial. Les protéines sont les principales molécules qui garantissent le fonctionnement des cellules. Ils sont responsables de réactions chimiques, de réguler les gènes, de transporter des molécules et de maintenir la structure cellulaire. Chaque cellule contient des milliers de protéines qui se forment de façon très dynamique. En fait, pour répondre aux besoins de la cellule actuelle ou aux stimuli externes, des protéines de différents types cellulaires sont constamment produites et détruites. Il est donc impératif de maintenir l'équilibre entre ces deux processus adverses pour assurer le bon fonctionnement de la cellule. Il n'est donc pas surprenant que beaucoup de processus de synthèse et de dégradation des protéines qui fouettent notre société soient à l'origine de nombreuses maladies. Parmi les maladies développées comme Alzheimer, Parkinson et Huntington, toutes très connues [1,2] (Figure 1).
Protéines: bien pliées ou finies.
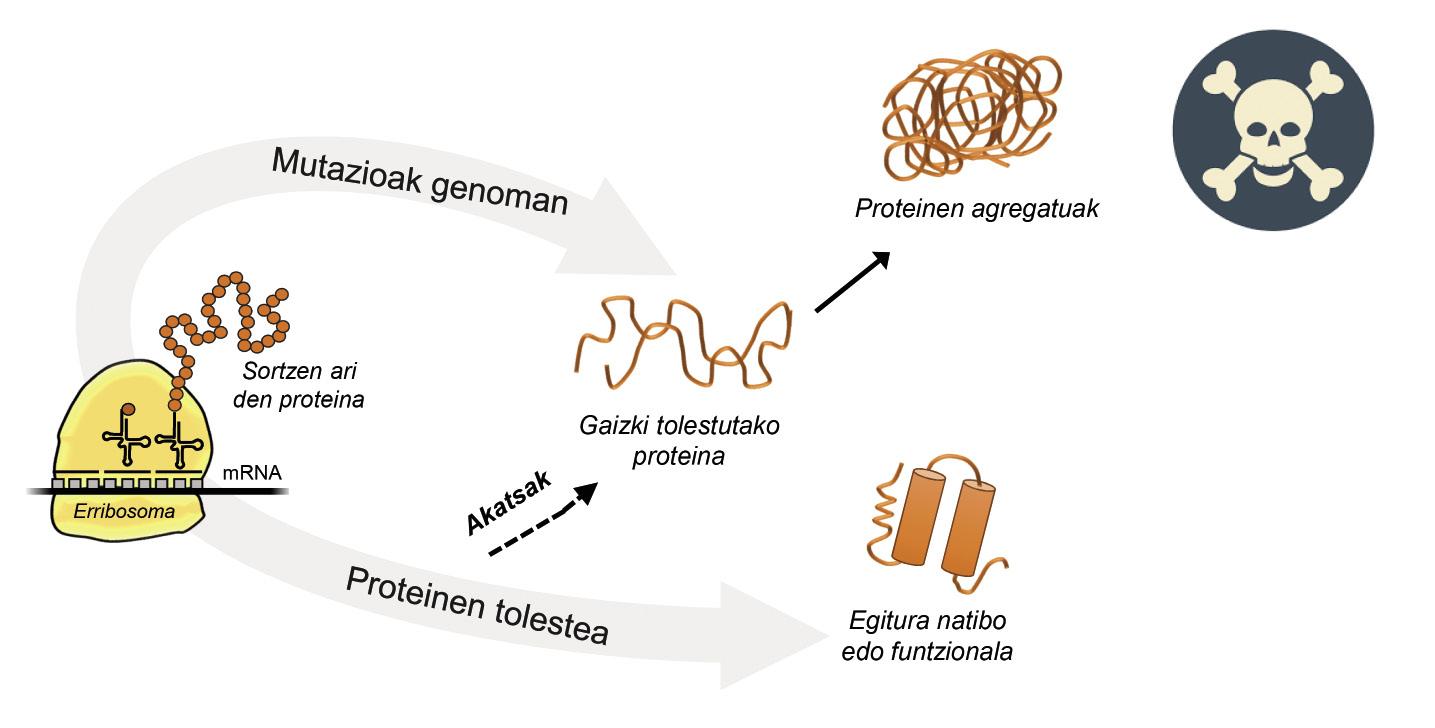
L'information pour la production de protéines est stockée dans les gènes. La chaîne ADN indique quand et quelle protéine sera générée. La structure d'une protéine qui est extraite de l'usine de production de protéines, c'est-à-dire des ribosomes, est équivalente à un collier de perles aux acides aminés. Cependant, cette protéine, pour être fonctionnelle, doit avoir une certaine structure tridimensionnelle. Dans la cellule on peut trouver 20 types d'acides aminés et la séquence d'acides aminés de chaque protéine détermine la structure fonctionnelle tridimensionnelle, c'est-à-dire la structure native de cette protéine. Les exigences cellulaires sont très spéciales, de sorte que les protéines ont souvent besoin de l'aide de protéines appelées chaperons moléculaires pour obtenir une structure native. Les humains ont plus de cent chapperons, dont chacun est responsable du processus de pliage de centaines de protéines ituelles. Lorsque l'activité des txaperons est endommagée par les mutations, le processus de pliage des protéines cibles n'est pas approprié, de sorte que ces protéines ne sont pas fonctionnelles. S'ils ne se dégradent pas, ils s'accumulent dans la cellule causant de graves dommages [4] (Figure 2).
Par exemple, les mutations du txaperon appelé HSPB1 provoquent une sclérose latérale amyotrophique et une neuropathie grave appelée Charcot Marie Tooth [5,6]. Et ceux qui ont le chaperon CHIP muté développent le syndrome de Gordon Holmes, un mal unique qui provoque l'hypogonadisme et des problèmes neurologiques.
Mais dans tous les cas où les protéines se plient mal et s'accumulent pathologiquement, la faute n'est pas des txaperos. Dans certains cas, en raison des mutations qui se produisent dans le génome, les protéines sont produites avec plus d'acides aminés que nécessaire, ce qui empêche d'obtenir la structure native correspondante (Figure 2).
Par exemple, les personnes touchées par la maladie de huntington ont une protéine de huntingtine défectueuse. En fait, lors de la mutation du gène HTT codant la huntingtine, la protéine produite contient plus d'acides aminés glutamine que nécessaire, c'est-à-dire contient trop d'exemplaires d'une perle particulière [4]. Les personnes touchées par la sclérose latérale multiple et la démence frontotemporelle présentent également une protéine C9orf72 défectueuse avec plus de paires d'acides aminés glycine que nécessaire, qui a une grande tendance à s'accumuler en neurones [7].
Cependant, la maladie la plus connue par l'accumulation de protéines est l'alzheimer, une maladie qui agite fortement la société actuelle. Comme les neurones meurent de l'hippocampe, les personnes malades perdent leurs capacités cognitives, en particulier la mémoire à court terme. Mais pourquoi les neurones meurent-ils ? Il y a de plus en plus de preuves que les accumulations de protéines tau et bêta-amyloïde mutées sont les principales responsables de la mort des neurones [6].
Protéasome: concasseur de protéines
Et la cellule n'a-t-elle pas des mécanismes pour dégrader des protéines mal pliées et éviter des agrégats pathologiques ? Oui, et plus d'une. Cependant, d'une part, compte tenu des exemples ci-dessus, il est évident que certaines accumulations de protéines sont capables de rompre avec ces mécanismes, et d'autre part, la machine elle-même de dégradation des protéines peut avoir des problèmes et empêcher le recyclage.
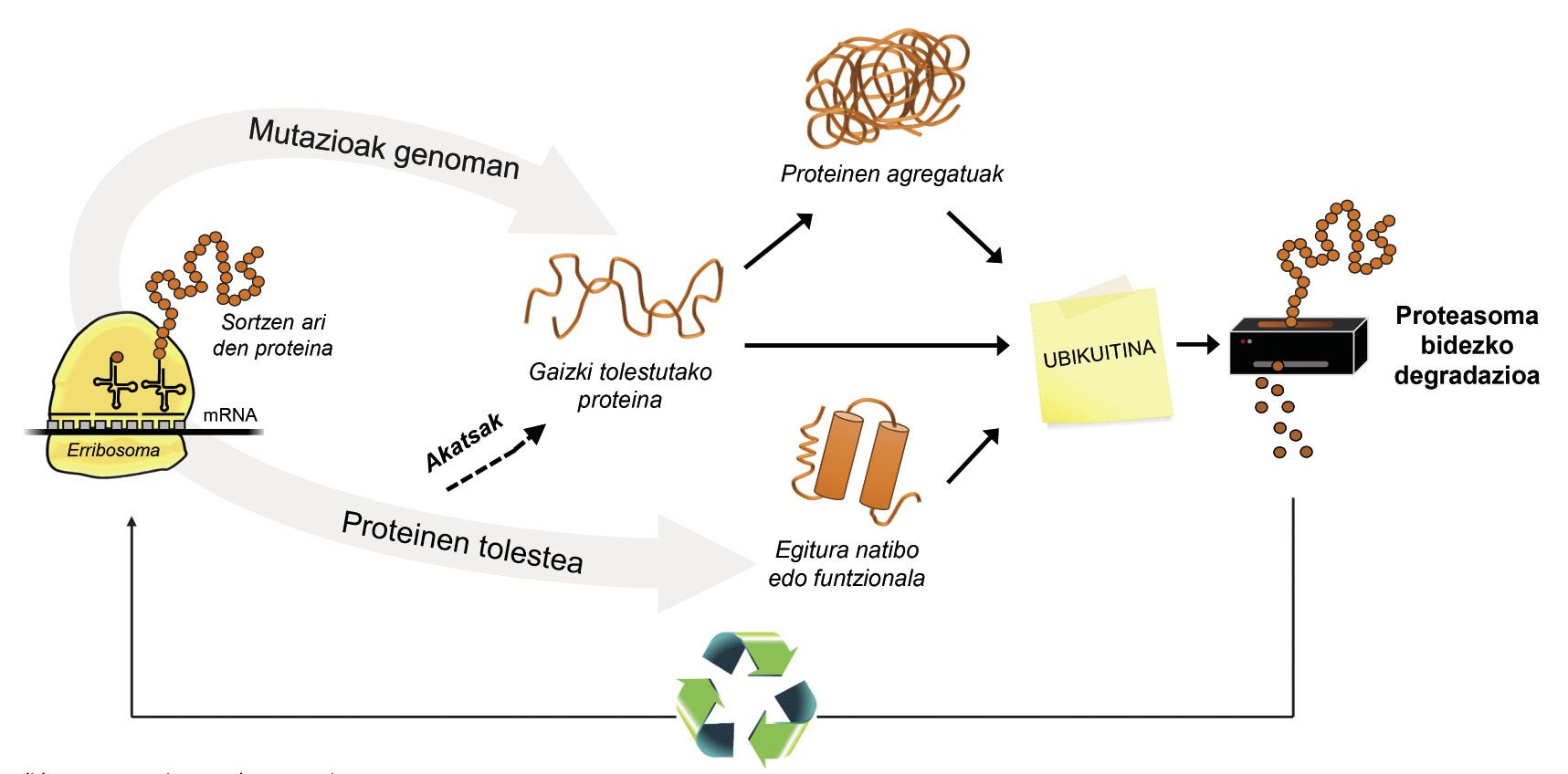
En plus des protéines mal pliées, la plupart des protéines non disponibles pour la cellule sont dégradées grâce à un complexe multiprotéique appelé protéasome. Mais parmi les milliers de protéines présentes dans la cellule, comment peut-on savoir ce qu'il faut dégrader ? Comment le protéasome percevt-il quelles protéines faut-il réduire ? Car ils ont adhéré une petite molécule appelée ubiquitine. Cette molécule fonctionne comme un post-it qui dit “vous devez aller à la protéasome à dégrader”. Proteasoma agit comme un broyeur de papier. Il réduit les protéines, c'est-à-dire rompt les relations entre les acides aminés. Parallèlement, il met à disposition de la cellule des acides aminés libres pour la production de nouvelles molécules [8]. On peut donc affirmer que le protéasome recycle des protéines (Figure 3).
Le processus de recyclage des protéines est très complexe et il suffit d'une erreur à n'importe quel point pour développer une maladie. Dans le processus de liaison de l'ubiquitine aux protéines qui vont être transférées à la protéasome, collaborent trois types d'enzymes: Enzymes E1, E2 et E3. Si ces enzymes étant mutées, le processus d'ubiquitination des protéines n'est pas correct, les protéines à dégrader ne sont pas détruites et accumulées dans la cellule. Cette toxicité se manifeste surtout dans les neurones hautement vulnérables, ce qui signifie que de nombreuses maladies neurologiques sont liées aux défauts qui se produisent dans ces enzymes [8].
L'enzyme UBA1 de type E1, par exemple, est liée à une maladie neuromusculaire exceptionnelle qui affecte uniquement les gars. Ces patients, en plus d'avoir une grande faiblesse musculaire, n'ont pas de réflexes, à savoir les muscles ne répondent pas correctement à des stimuli externes. Ceux qui ont muté l'enzyme UBE2A type E2 développent le syndrome de carence UBE2A, une maladie neurologique atypique. Cette maladie est également sujette uniquement aux hommes, les symptômes les plus fréquents étant la déficience mentale, les troubles cutanés, de l'appareil reproducteur et urinaire et l'absence de parole.
Parmi les trois enzymes qui effectuent l'ubiquitination des protéines, les plus abondantes sont les E3. Ainsi, il existe une enzyme E3 mutée derrière la plupart des maladies neurologiques développées à la suite d'une ubichitination défectueuse. Par exemple, ceux qui souffrent de l'autisme familial et de l'autisme sporadique, classés dans les troubles du spectre de l'autisme, ont muté les enzymes UBE3B et UBE3C type E3, respectivement. Au contraire, les personnes atteintes d'UBE3A défectueuse développent le syndrome d'Angelman lié à l'autisme [9].
En plus des maladies liées à l'autisme, les E3 mutés peuvent causer d'autres maladies neurologiques très différentes. Par exemple, ceux qui ont muté l'enzyme E3 appelée gigaxonine développent la neuropathie des axons géants. Dans des conditions saines, la gigaxonine stabilise les neurofilaments des bras neuronaux, c'est-à-dire des axons, en conditionnant leur structure et leur taille. Cependant, lorsque la gigaxonine se déplace, il devient non fonctionnel et non vascuitine neurofilaments des axons. Cela ne dégrade pas les neurofilaments et rend les axons des neurones plus grands et plus longs que nécessaire. En conséquence, la plupart des patients souffrant de cette neuropathie ont des problèmes pour marcher et, en général, pour coordonner les mouvements [9].
Après le récent syndrome de Tenorio se trouve une enzyme E3, dans ce cas appelée RNF125. Les personnes touchées par le syndrome de Tenorio ont un handicap intellectuel. En outre, ce type de patients sont caractérisés par une croissance excessive, en particulier le développement excessif du crâne. Le contraire arrive à ceux qui ont muté le TRIM36, à qui manque une grande partie du crâne et du cerveau [9].

Conclusions
Il est évident qu'un déséquilibre dans le cycle de synthèse et de dégradation des protéines peut produire des effets vraiment nocifs pour les cellules et les organismes vivants en général. D'une part, des protéines mal pliées par des mutations, accumulées comme sacs à ordures inutilisables, peuvent causer la mort des cellules. Par ailleurs, la détérioration des dispositifs moléculaires responsables de la dégradation et du recyclage des protéines produit des accumulations toxiques de protéines. De manière analogue à l'accumulation de déchets si l'activité d'une installation de recyclage des déchets est annulée.
Nous savons que l'un des principaux défis de la société actuelle est la bonne gestion des déchets. En effet, si le recyclage des déchets n'est pas garanti, nous couvrirons nos océans et nos champs de déchets en quelques années et la vie deviendra insoutenable. Dans le monde microscopique les cellules ont un défi semblable quand elles ont des problèmes pour gérer des protéines non fonctionnelles. Pour garantir votre santé, il est indispensable d'éliminer les protéines toxiques.
Donc, si la prochaine fois vous oubliez de recycler les déchets de votre maison, rappelez-vous le désastre que cela impliquerait pour une cellule. En fait, le recyclage des déchets que nous générons les humains et le recyclage des protéines qui s'accumulent dans les cellules sont des actions similaires, à grande et petite échelle. Une clé pour que nous soyons tous en bonne santé.
Bibliographie Bibliographie
Zu idazle
Zientzia aldizkaria

- Babesleak
-

-

Elhuyar
Nor gara | Kontaktua |
Publizitatea
| Laguntza
Pribatutasun politika | Cookien politika
ISSN-2603-6614 Elhuyar