

Machines pour la prévention des maladies neurodégénératives
Pliage de protéines et de chapperons moléculaires
Les protéines sont des composants fondamentaux de la vie, car elles garantissent le fonctionnement des cellules. Ces molécules, formées par un petit alphabet de vingt acides aminés, s'occupent au maximum des fonctions qui ont lieu dans la cellule, des réactions chimiques aux fonctions structurelles. La collection de protéines – protéome – qui se trouve dans un organisme à un moment donné est très variable et presque infinie, bien que toutes les informations pour construire la collection soient codifiées dans les gènes.
Le dogme principal de la biologie moléculaire explique le transfert d'information de l'ADN aux protéines, étant l'ARN médiateur. L'expression des gènes commence à partir de la transcription, où, à partir de la double molécule d'ADN, se produit la chaîne auxiliaire d'ARN, qui sera une copie de cette information génétique concrète. Dans la deuxième étape, l'ARN messager retourne à la machine appelée ribosome, obtenant la protéine. La protéine nouvellement synthétisée doit assimiler une structure tridimensionnelle appropriée pour être fonctionnellement active, mais dans les cellules ce processus n'est pas toujours spontané.
Les chaperons moléculaires sont essentiels dans les cellules, car ils contrôlent la qualité des protéines, garantissant ainsi l'homéostasie des protéines2. Et c'est que beaucoup de protéines ont besoin de l'aide des txaperons pour obtenir leur structure nativ3. Sans votre aide, des problèmes peuvent se produire dans le processus de pliage de la protéine et former des intermédiaires mal pliés. Ces intermédiaires affleureront les zones adhésives qui devraient être dans le noyau de la protéine et tendront à son agrégation4. En outre, l'agrégation d'un type de protéines peut influencer l'agrégation d'autres espèces de protéines, permettant ce qui est connu comme coagrégation.
Le processus de pliage des protéines nouvellement synthétisées n'est pas la seule voie pour générer les premiers intermédiaires des arides. En fait, les protéines qui possèdent déjà une structure tridimensionnelle appropriée (structure native) peuvent également subir des décolorations et former des intermédiaires réactifs. Les principales causes de cette déstabilisation sont liées aux mutations ou au stress cellulaire, qui est plus élevé chez les personnes âgées, car les mécanismes d'homéostasie des protéines sont plus obsolètes.
En fait, dans une situation normale, face à tout stress cellulaire, les chaperons moléculaires le perçoivent et répondent rapidement, maintenant ainsi l'homéostasie des protéines. Cependant, lorsque des situations de stress sont imposées ou chroniques, comme les mutations, il est plus difficile de garantir la qualité des protéines des cellules6. Dans ces circonstances, la présence de txaperons moléculaires est moindre pour faire face aux problèmes de flexion des protéines (en raison de leur moindre expression ou de la saturation du système pour contrer le stress cellulaire), augmentant la quantité de protéines endommagées et mal pliées7.

L'agrégation entrave le fonctionnement cellulaire et provoque l'apparition de maladies liées à la flexion des protéines comme la maladie d'Alzheimer, le parkinson ou d'autres maladies neurodégénératives8. En fait, plusieurs maladies neurodégénératives présentent une caractéristique commune : l'accumulation de protéines agrégées de structure amyloïde présentes dans le cerveau.
Agrégation et effets des arides
Les processus d'agrégation de protéines dans ces maladies sont lents et organisés. Actuellement, le modèle le plus accepté pour expliquer la formation de structures amyloïdogènes est la polymérisation en nucléation. Selon lui, le processus d'agrégation présente une cinétique caractéristique qui peut être divisée en deux phases distinctes: lag ou nucléaire et exponentielle ou d'étirement.
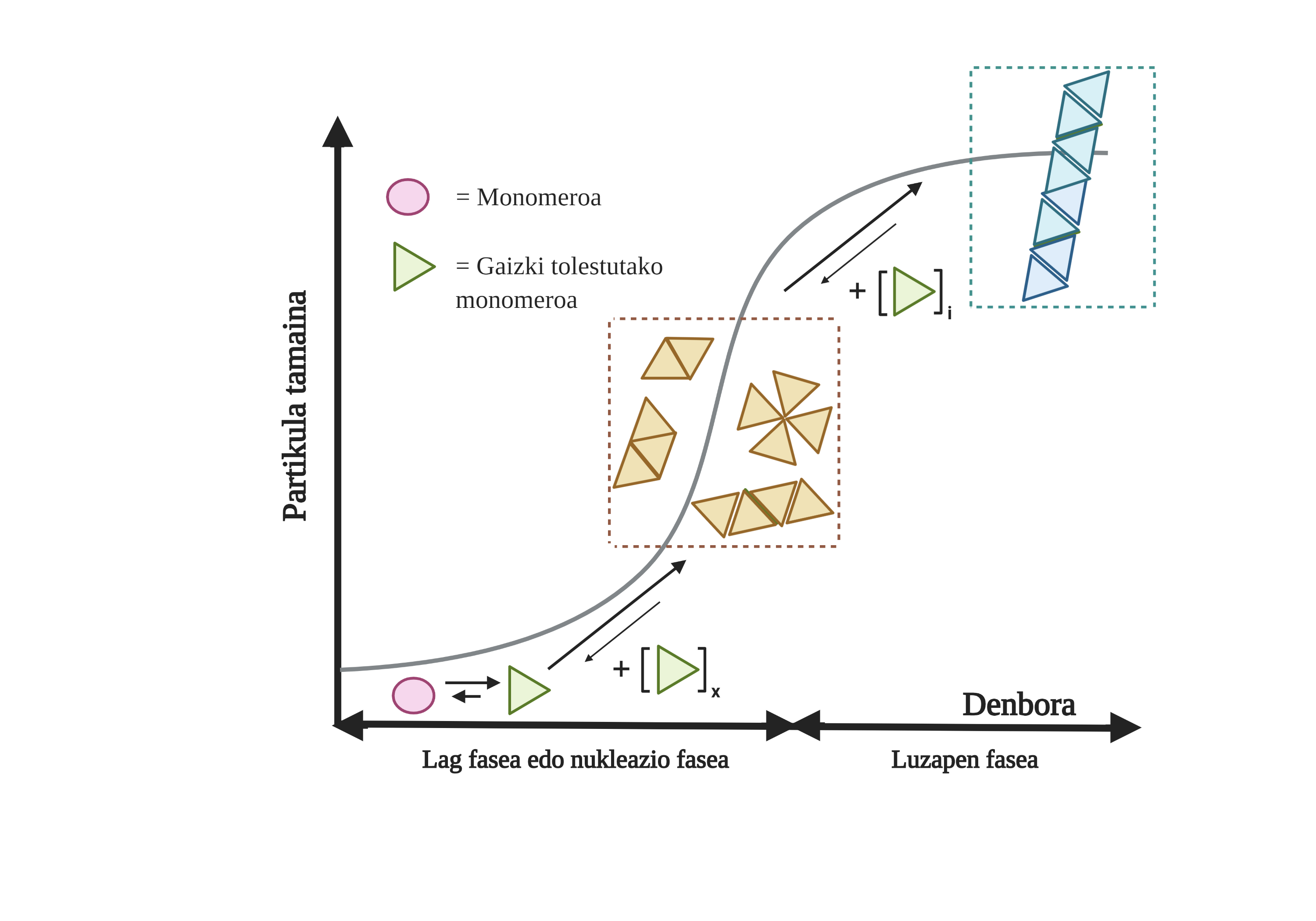
Dans la phase de Lag sont créés les premiers intermédiaires appelés graines ou noyau, les réactifs. Dans ces structures se forment de petites ou oligomériques agrégées de protéines mal pliées qui se sont unies entre elles et qui commencent la deuxième phase. Ces petits agrégats ou oligomères accumulés en phase exponentielle attirent plus de protéines formant des agrégats toujours plus grands jusqu'à former des fibres amyloïdes 10.
Aujourd'hui, il n'est pas tout à fait clair quel est le rôle des arides, mais il semble que les intermédiaires d'agrégation sont les plus toxiques pour la cellule. Selon cette hypothèse, le processus d'agrégation serait donc une méthode de protection cellulaire par rapport aux intermédiaires toxiques. Dans tous les cas, on sait que les accumulations d'espèces amyloïdes sont toxiques et que leur accumulation produit une apoptose neuronale ou une mort cellulaire programmée dans le cerveau.
La mort naturelle, précise et programmée des neurones, l'apoptose neuronale, est un processus indispensable pour la maturation du système nerveux central. Cependant, une fois que le système nerveux central est bien développé, la plupart des neurones adultes resteront tout au long de la vie de l'organisme, car le taux d'apoptose des neurones est très faible. L'apoptose précoce des neurones ou l'apoptose aberrante mal réglementée provoque l'apparition de maladies neurodégénératives. Compte tenu de la zone du cerveau où se produit l'accumulation d'espèces amyloïdogènes et l'apoptose conséquente des neurones, une maladie néodégénérative différente se développera. Par exemple, la perte de neurones d'hippocampe est liée à la maladie d'Alzheimer, alors que la diminution des neurones dopaminergiques dans le noir de substance est liée au parkinson 14.
Le rôle des txaperons moléculaires devant les intermédiaires toxiques
En plus de contribuer au processus de pliage de protéines nouvellement synthétisées, les txaperons moléculaires participent également à la repliation d'intermédiaires mal pliés, permettant la disparition d'intermédiaires réactifs. Par conséquent, les txaperons moléculaires aident à la fois dans la prévention et la repliation, en évitant la formation d'intermédiaires toxiques ou en garantissant la libération et la virilisation des protéines présentes dans les oligomères lorsque la formation d'intermédiaires est inévitable.
Pour permettre la régénération des protéines mal pliées ou agrégées, il est nécessaire de coordonner trois familles de chapperons moléculaires: Hsp70, Hsp40 et Hsp110. Parmi les trois forment le système Hsp70, qui, en utilisant l'énergie mécanique libérée de l'hydrolyse de l'ATP, permet aux protéines mal pliées d'obtenir une structure native grâce au processus appelé cycle de l'ATP (Figure 3). La protéine Hsp70 se compose de deux domaines : le domaine d'union du nucléotide (NBD), dans lequel nous aurons des nucléotides ATP ou ADP, et le domaine d'union du substrat (SBD), qui selon le nucléotide associé au NBD, sera en formage ouvert ou fermé.
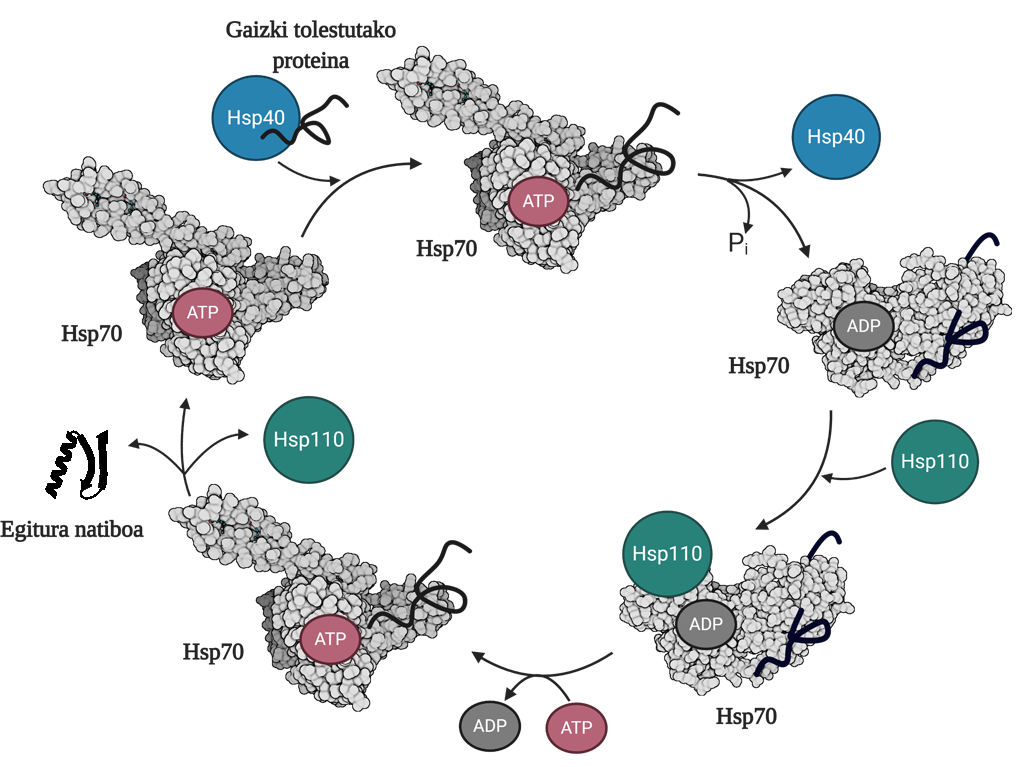
La protéine Hsp40 affectera un médiateur mal plié et transférera au chaperon Hsp70 qui est attaché à l'ATP, dont le domaine SBD sera en conformation ouverte en présence de l'ATP. Par stimulation du substrat et de la protéine Hsp40, le Hsp70 hydrolyse d'ATP, restant l'ADP soumis au Hsp70. Dans cette situation, le Hsp40a sera libéré du complexe et le substrat sera piégé dans le SBD du Hsp70, car en présence de l'ADP ce domaine acquerra une conformation fermée. À ce moment-là, le cycle comprendra le Hsp110 ou l'échangeur de nucléotides (NEF) et échangera l'ADP contenant le Hsp70 avec un nouvel ATP. En raison de la présence de l'ATP, le SBD passera à nouveau au formage ouvert et libérera le substrat au milieu. De cette façon, le substrat aura une autre possibilité de bien se plier, normalement il sera en mesure d'obtenir une structure native et la protéine Hsp70 sera recyclée pour un prochain cycle 10.
La fonction de désagrégation/repliage du système Hsp70 est donc indispensable pour éviter l'accumulation d'intermédiaires toxiques, car en plus d'éviter la génération de ces toxiques, il est capable de réaliser la désagrégation d'oligomères déjà constitués. Le fonctionnement efficace de ce système permet de prévenir les maladies liées à des problèmes de pliage tels que le parkinson, l'alzheimer ou la sclérose latérale amyotrophique.
Cancer et apoptose
L'une des principales caractéristiques du cancer est l'immortalité des cellules cancéreuses. La division rapide des cellules cancéreuses et la résistance à l'apoptose sont les principaux responsables de l'augmentation de la tumeur. Les cellules cancéreuses utilisent différentes voies pour éviter l'apoptose, principalement l'inhibition des signaux pro-apoptotiques et l'augmentation des stimuli anti-apoptotiques. Ils parviennent ainsi à maintenir les cellules tumorales, malgré un stress élevé, sans entrer dans l'apoptose. Le système Hsp70 étant un mécanisme anti-stress, la surexpression du chaperon Hsp70 est très fréquente dans les cellules cancéreuses, ce qui favorise la résistance à l'apoponyon16.
Le système Hsp70 comme cible thérapeutique au cancer
Étant donné l'importance du Hsp70 dans les cellules tumorales, il semble qu'il peut être une cible thérapeutique appropriée pour réduire la résistance des cellules cancéreuses aux médicaments et donc diminuer les tumeurs 17. Des études montrent que la diminution de l'expression du Hsp70 est toxique dans les cellules cancéreuses, mais n'est pas évidente dans les cellules non modifiées18. L'inhibition du Hsp70 affecte davantage les cellules tumorales, qui auront la priorité de mort par rapport aux cellules normales. En fait, le phénotype (constamment stressé) de la cellule tumorale dépend de la fonction protectrice du Hsp70 pour combattre la cytotoxicité et donc éviter l'apopto19. Hrrn a considérablement élargi le champ de recherche du système Hsp70; l'utilisation du chaperon Hsp70 comme cible a un grand avenir, car il peut être utile non seulement pour le traitement des maladies neurodégénératives, mais aussi pour les traitements du cancer.
Références
1- Crick F. 1970. “Dogme central de la biologie moléculaire.” Nature, 227(5258): 561–63.
2- Fin B, Weissman, J. et Horwich, A. 2006. “Moléculaire Chaperons and Protein Quality Control.” Cell, 125(3): 443–51.
3- Gao X, Carroni M. Nussbaum-Krammer C, Mogk A., Nillegoda N.B., Szlachcic A. Guilbride D.L. H.R. Saibil, Mayer M.P. et B. Finale. 2015 “Human Hsp70 Disaggregase Reverses Parkinson’s-Linked ?-Synuclein Amyloid Fibrils.” Moléculaire Cell, 59(5): 781–93.
4- U.T.E. 1996. “Moléculaire Chaperons in Cellular Protein Folding.” Nature, 381(6583): 571–79.
5- Morimoto R.I. 2008. “Proteotoxic stress and induisible chaperone networks in neurodegenerative disease and aging.” Gènes Developement, 22(11): 1427–1438.
6- Radwan M., Wood R.J. Sui X, et Hatters D.M. 2017 “When proteostasis goes bad: protein aggregation in the cell.” IUBMB Life, 69, 49–54.7
7- Stroo E., Koopman M., O.A.F. Nollen et Mata-Cabana A. 2017 “Cellular Regulation of Amyloid Formation in Aging and Disease.” Forntiers in Neuroscience, 11: 64.
8- Stetler R.A., Et., Zhang W., C. O. Liou, Y. Gao, Cao G. et Chen J. 2010 “Heat Shock Proteins: Cellular and Molecular Mechanisms in the Central Nervous System.” Progress in Neurobiology, 92(2): 184-211.
9- GoM. et Spillantini M.G. 2006. “A century of Alzheimer’s disease.” Science, 314, 777–781.
10- Chaari A. 2019. “Moléculaire chaperons biochemistry and role in neurodegenerative diseases.” International Journal of Biological Macromolecules, 131:396-411.
11- Hartl F.U., et Hayer-Hartl M. 2009. “Converging concepts of protein folding in vitro and in vivo.” Nature Structural and Molecular Biology, 16, 574–581.
12- Awasthi A., Matsunaga Y et Yamada T. 2005 “Amyloid-beta causes apoptosis of neuronal cells via caspaso cascade, which can be prevented by amyloid-beta-dérived short peptides.” Experimental Neurology, 196(2): 282-289.
13- Okouchi M, Ekshyyan O., Maracine M. et Aw T.Y. 2007 “Apoptose neuronale en neurodegeneration.” Antioxydants Redox Signaling, 9(8): 1059-1096.
14- Chi H., Chang H.Y et Sang T.K. 2018. “Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases.” International Journal of Molecular Sciences, 19(10): 3082.
15- Hanahan D. et Weinberg R.A. 2000. “The Hallmarks of Cancer.” Cell, 100(1): 57-70.
16- D. Brusa, C. Migliore, Garetto S., Simone M. et Matera L. 2009. “Immunogenicity of 56 degrees C and uvc-treated prostate cancer is associated with release of HSP70 and HMGB1 from necrotic cells.” Prostate, 69: 1343–1352.
17- S. Gurbuxani, Bruey J.M. Fromentin A., C. Larmonier, Parcellier A., Jaattel M., Martin F., Solary E. et Garrido C. 2001. “Selective depletion of induisible HSP70 enhances immunogenicity of rat colon cancer cells.” Oncogene, 20: 7478–7485.
18- Garrido C., Schmitt E., Cande C., Vahsen N., Parcellier A. et Kroemer G. 2003. “HSP27 and HSP70: potentially oncogenic apoptosis inhibitors.” Cell Cycle, 2: 579–584.
19- Kumar S., J. Strokes, Singh U.P., S.G. Gunn, Acharya A., Manne U. et Mishra M. 2016 “Target Hsp70: A possible therapy for cancer.” Cancer Letters, 374(1): 156-166.
Zu idazle
Zientzia aldizkaria

