

Maquinaria para la prevención de enfermedades neurodegenerativas
Plegado de proteínas y chapperones moleculares
Las proteínas son componentes básicos de la vida, ya que garantizan el funcionamiento de las células. Estas moléculas, formadas por un pequeño alfabeto de veinte aminoácidos, se ocupan como máximo de las funciones que tienen lugar en la célula, desde reacciones químicas hasta funciones estructurales. La colección de proteínas –proteoma– que hay en un organismo en un momento dado es muy variable y casi infinita, aunque toda la información para construir la colección está codificada en los genes.
El dogma principal de la biología molecular explica la transferencia de información del ADN a las proteínas, siendo el ARN mediador. La expresión de los genes se inicia a partir de la transcripción, donde, a partir de la doble molécula de ADN, se produce la cadena auxiliar de ARN, que será una copia de esa información genética concreta. En el segundo paso, el ARN mensajero vuelve a la maquinaria denominada ribosoma, obteniendo la proteína. La proteína recién sintetizada deberá asimilar una estructura tridimensional adecuada para ser funcionalmente activa, pero en las células este proceso no siempre es espontáneo.
Los chaperones moleculares son esenciales en las células, ya que se encargan del control de calidad de las proteínas, garantizando así la homeostasis de las proteinas2. Y es que muchas proteínas necesitan la ayuda de los txaperones para conseguir su estructura nativ3. Sin su ayuda pueden producirse problemas en el proceso de plegado de la proteína y formar intermediarios mal plegados. Estos intermediarios aflorarán las zonas adhesivas que deberían estar en el núcleo de la proteína y tenderán a su agregación4. Además, la agregación de un tipo de proteínas puede influir en la agregación de otras especies de proteínas, permitiendo lo que se conoce como coagregación.
El proceso de plegado de proteínas recién sintetizadas no es la única vía para generar los primeros intermediarios de los áridos. De hecho, las proteínas que ya poseen una estructura tridimensional adecuada (estructura nativa) también pueden sufrir destolaciones y formar intermediarios reactivos. Las principales causas de esta desestabilización están relacionadas con mutaciones o estrés celular, que es mayor en los mayores, ya que los mecanismos de homeostasis de las proteínas son más obsoletos.
De hecho, en una situación normal, ante cualquier estrés celular, los chaperones moleculares lo perciben y responden rápidamente, manteniendo así la homeostasis de las proteínas. Sin embargo, cuando se van imponiendo o se cronifican situaciones de estrés, como las mutaciones, es más difícil garantizar la calidad de las proteínas de las celulas6. En estas circunstancias, la presencia de txaperones moleculares es menor para hacer frente a los problemas de flexión de las proteínas (debido a su menor expresión o a la saturación del sistema por contrarrestar el estrés celular), aumentando la cantidad de proteínas dañadas y mal plegadas7.

La agregación dificulta el funcionamiento celular y provoca la aparición de enfermedades relacionadas con la flexión de proteínas como el alzheimer, el parkinson u otras enfermedades neurodegenerativas8. De hecho, varias enfermedades neurodegenerativas presentan una característica común: la acumulación de proteínas agregadas de estructura amiloide presentes en el cerebro.
Agregación y efectos de los áridos
Los procesos de agregación de proteínas en estas enfermedades son lentos y organizados. En la actualidad, el modelo más aceptado para explicar la formación de estructuras amiloidogénicas es la polimerización en nucleación. Según él, el proceso de agregación presenta una cinética característica que puede dividirse en dos fases diferenciadas: lag o nuclear y exponencial o de estiramiento.
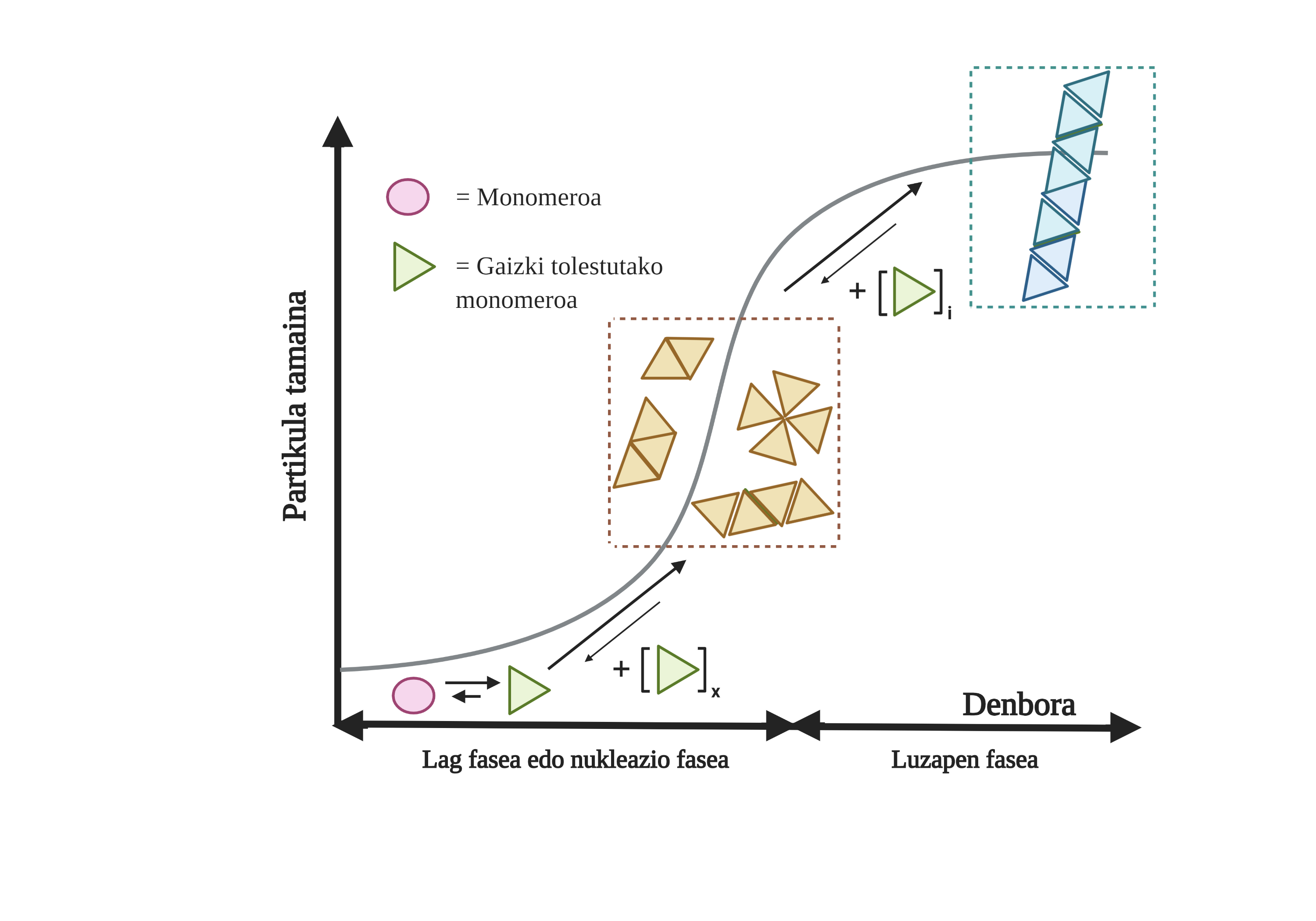
En la fase de Lag se crean los primeros intermediarios denominados semilla o núcleo, los reactivos. En estas estructuras se forman pequeños o oligoméricos agregados de proteínas mal plegadas que se han unido entre sí y que comienzan la segunda fase. Estos pequeños agregados o oligómeros acumulados en fase exponencial atraen más proteínas formando agregados cada vez más grandes hasta formar fibras amiloides 10.
En la actualidad no está del todo claro cuál es el papel de los áridos, pero parece que los intermediarios de la agregación son los más tóxicos para la célula. Según esta hipótesis, el proceso de agregación sería, por tanto, un método de protección celular frente a intermediarios tóxicos. En cualquier caso, se sabe que las acumulaciones de especies amiloides son tóxicas y que su acumulación produce una apoptosis neuronal o muerte celular programada en el cerebro.
La muerte natural, precisa y programada de las neuronas, la apoptosis neuronal, es un proceso imprescindible para la maduración del sistema nervioso central. Sin embargo, una vez que el sistema nervioso central esté bien desarrollado, la mayoría de las neuronas adultas se mantendrán a lo largo de toda la vida del organismo, ya que la tasa de apoptosis de las neuronas es muy baja. La apoptosis precoz de las neuronas o la apoptosis aberrante mal regulada provoca la aparición de enfermedades neurodegenerativas. Teniendo en cuenta en qué zona del cerebro se produce la acumulación de especies amiloidogénicas y la consiguiente apoptosis de las neuronas, se desarrollará una enfermedad neodegenerativa diferente. Por ejemplo, la pérdida de neuronas del hipocampo está relacionada con el alzheimer, mientras que la disminución de neuronas dopaminérgicas en el negro de sustancia está relacionada con el parkinson 14.
El papel de los txaperones moleculares ante los intermediarios tóxicos
Además de contribuir al proceso de plegado de proteínas recién sintetizadas, los txaperones moleculares también participan en la replegación de intermediarios mal plegados, permitiendo la desaparición de intermediarios reactivos. Por lo tanto, los txaperones moleculares ayudan tanto en la prevención como en la replegación, evitando la formación de intermediarios tóxicos o garantizando la liberación y virilización de proteínas presentes en los oligómeros cuando la formación de intermediarios es inevitable.
Para permitir la regeneración de proteínas mal plegadas o agregadas es necesario coordinar tres familias de chapperones moleculares: Hsp70, Hsp40 y Hsp110. Entre los tres forman el sistema Hsp70, que, utilizando la energía mecánica liberada de la hidrólisis del ATP, permite a las proteínas mal dobladas obtener una estructura nativa gracias al proceso denominado ciclo del ATP (Figura 3). La proteína Hsp70 consta de dos dominios: el dominio de unión del nucleótido (NBD), en el que tendremos nucleótidos ATP o ADP, y el dominio de unión del sustrato (SBD), que dependiendo del nucleótido asociado al NBD, estará en conformado abierto o cerrado.
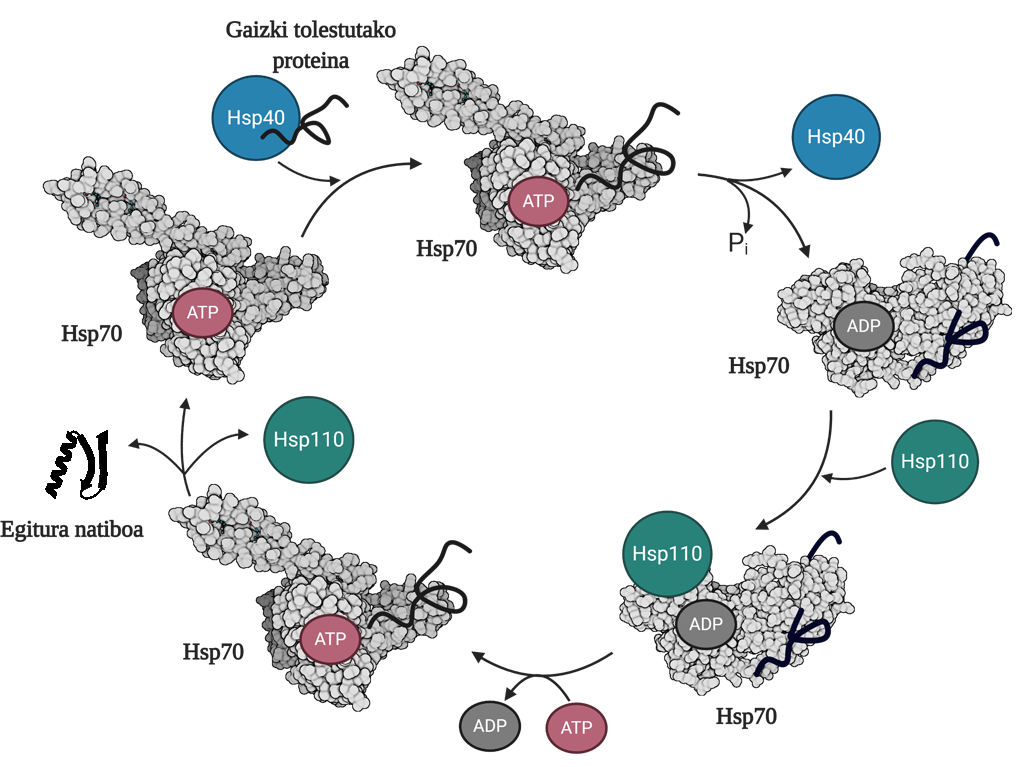
La proteína Hsp40 afectará a un mediador mal plegado y transferirá al chaperón Hsp70 que está unido al ATP, cuyo dominio SBD estará en conformación abierta en presencia del ATP. Por estimulación del sustrato y de la proteína Hsp40, el Hsp70 hidrólisis del ATP, quedando el ADP sujeto al Hsp70. En esta situación, el Hsp40a se liberará del complejo y el sustrato quedará atrapado en el SBD del Hsp70, ya que en presencia del ADP este dominio adquirirá una conformación cerrada. En ese momento, el ciclo incluirá el Hsp110 o el intercambiador de nucleótidos (NEF) y intercambiará el ADP que contiene el Hsp70 con un nuevo ATP. Debido a la presencia del ATP, el SBD volverá a pasar al conformado abierto y liberará el sustrato al medio. De esta forma, el sustrato tendrá otra posibilidad de plegarse bien, normalmente será capaz de obtener una estructura nativa y la proteína Hsp70 será reciclada para un próximo ciclo 10.
La función de desagregación/replegado del sistema Hsp70 es, por tanto, imprescindible para evitar la acumulación de intermediarios tóxicos, ya que además de evitar la generación de estos tóxicos, es capaz de llevar a cabo la desagregación de oligómeros ya constituidos. El funcionamiento eficiente de este sistema conlleva la capacidad de prevenir enfermedades relacionadas con problemas de plegamiento como el parkinson, el alzheimer o la esclerosis lateral amiotrófica.
Cáncer y apoptosis
Una de las principales características del cáncer es la inmortalidad de las células cancerosas. La rápida división de las células cancerosas y la resistencia a la apoptosis son los principales responsables del aumento del tumor. Las células cancerígenas utilizan diferentes vías para evitar la apoptosis, principalmente inhibición de las señales pro-apoptóticas y aumento de los estímulos anti-apoptóticos. De esta forma consiguen mantener las células tumorales, a pesar de sufrir un elevado estrés, sin entrar en la apoptosis. Siendo el sistema Hsp70 un mecanismo anti-estrés, en las células cancerígenas es muy frecuente la sobreexpresión del chaperón Hsp70, lo que favorece la resistencia a la apoptosión16.
El sistema Hsp70 como diana terapéutica al cáncer
Dada la importancia que tiene el Hsp70 en las células tumorales, parece que puede ser una diana terapéutica adecuada para reducir la resistencia de las células cancerígenas a los fármacos y por tanto disminuir los tumores 17. Hay estudios que demuestran que la disminución de la expresión del Hsp70 es tóxica en las células cancerígenas, pero no es evidente en las células no modificadas18. La inhibición del Hsp70 afecta más a las células tumorales, que tendrán prioridad de muerte respecto a las células normales. De hecho, el fenotipo (constantemente estresado) de la célula tumoral depende de la función protectora del Hsp70 para combatir la citotoxicidad y por tanto evitar la apopto19. Hrrn ha ampliado notablemente el campo de investigación del sistema Hsp70; el uso del chaperón Hsp70 como diana tiene un gran futuro, ya que puede ser útil no sólo para el tratamiento de enfermedades neurodegenerativas, sino también para tratamientos de cáncer.
Referencias
1- Crick F. 1970. “Central Dogma of Molecular Biology.” Nature, 227(5258): 561–63.
2- Fin B, Weissman, J. y Horwich, A. 2006. “Molecular Chaperones and Protein Quality Control.” Cell, 125(3): 443–51.
3- Gao X, Carroni M. Nussbaum-Krammer C, Mogk A., Nillegoda N.B., Szlachcic A. Guilbride D.L. H.R. Saibil, Mayer M.P. y B. Final. 2015 “Human Hsp70 Disaggregase Reverses Parkinson’s-Linked ?-Synuclein Amyloid Fibrils.” Molecular Cell, 59(5): 781–93.
4- U.T.E. 1996. “Molecular Chaperones in Cellular Protein Folding.” Nature, 381(6583): 571–79.
5- Morimoto R.I. 2008. “Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging.” Genes & Developement, 22(11): 1427–1438.
6- Radwan M., Wood R.J. Sui X, y Hatters D.M. 2017 “When proteostasis goes bad: protein aggregation in the cell.” IUBMB Life, 69, 49–54.7
7- Stroo E., Koopman M., O.A.F. Nollen y Mata-Cabana A. 2017 “Cellular Regulation of Amyloid Formation in Aging and Disease.” Forntiers in Neuroscience, 11: 64.
8- Stetler R.A., Y., Zhang W., C. O. Liou, Y. Gao, Cao G. y Chen J. 2010 “Heat Shock Proteins: Cellular and Molecular Mechanisms in the Central Nervous System.” Progress in Neurobiology, 92(2): 184–211.
9- GoM. y Spillantini M.G. 2006. “A century of Alzheimer’s disease.” Science, 314, 777–781.
10- Chaari A. 2019. “Molecular chaperones biochemistry and role in neurodegenerative diseases.” International Journal of Biological Macromolecules, 131:396-411.
11- Hartl F.U., y Hayer-Hartl M. 2009. “Converging concepts of protein folding in vitro and in vivo.” Nature Structural and Molecular Biology, 16, 574–581.
12- Awasthi A., Matsunaga Y y Yamada T. 2005 “Amyloid-beta causes apoptosis of neuronal cells via caspase cascade, which can be prevented by amyloid-beta-derived short peptides.” Experimental Neurology, 196(2): 282-289.
13- Okouchi M, Ekshyyan O., Maracine M. y Aw T.Y. 2007 “Neuronal Apoptosis in Neurodegeneration.” Antioxidants & Redox Signaling, 9(8): 1059-1096.
14- Chi H., Chang H.Y y Sang T.K. 2018. “Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases.” International Journal of Molecular Sciences, 19(10): 3082.
15- Hanahan D. y Weinberg R.A. 2000. “The Hallmarks of Cancer.” Cell, 100(1): 57-70.
16- D. Brusa, C. Migliore, Garetto S., Simone M. y Matera L. 2009. “Inmunogenicity of 56 degrees C and uvc-treated prostate cancer is associated with release of HSP70 and HMGB1 from necrotic cells.” Prostate, 69: 1343–1352.
17- S. Gurbuxani, Bruey J.M. Fromentin A., C. Larmonier, Parcellier A., Jaattel M., Martín F., Solary E. y Garrido C. 2001. “Selective depletion of inducible HSP70 enhances inmunogenicity of rat colon cancer cells.” Oncogene, 20: 7478–7485.
18- Garrido C., Schmitt E., Cande C., Vahsen N., Parcellier A. y Kroemer G. 2003. “HSP27 and HSP70: potentially oncogenic apoptosis inhibitors.” Cell Cycle, 2: 579–584.
19- Kumar S., J. Strokes, Singh U.P., S.G. Gunn, Acharya A., Manne U. y Mishra M. 2016 “Target Hsp70: A possible therapy for cancer.” Cancer Letters, 374(1): 156-166.
Zu idazle
Zientzia aldizkaria

