

Outils de diagnostic génétique des abeilles de miel européennes

Le nom mellifera ou abeille de miel est aujourd'hui le pollinisateur le plus important au monde, à la fois écologique et économique, et est fréquemment utilisé pour la production de miel et la pollinisation des cultures forestières. Cependant, ces abeilles sont menacées par divers facteurs. Parmi elles, la réduction de la diversité génétique et la perte de sous-espèces locales adaptées au milieu ont une influence notable, tant par la croissance et l'importation commerciale de reines à grande échelle que par l'apiculture migratoire à longue distance. Pour garantir la santé de la population mondiale, il faut assurer la conservation des sous-espèces locales et maintenir leur héritage naturel et leur capacité d'adaptation. L'Europe est riche du patrimoine génétique des abeilles de miel (représentées par au moins 13 sous-espèces de 4 lignées évolutives de 5 lignées définies dans l'espèce) et a été une région fondamentale pour la conservation et le développement d'outils qui la facilitent. C'est pourquoi, dans le cadre du projet européen SmartBees a été développé une puce d'ADN qui permettra l'identification de sous-espèces d'abeilles de miel européennes. Diagnostic.- Cet outil facilitera et stimulera les activités visant à préserver la conservation durable, le commerce contrôlé des reines, la vérification des produits apiculteurs et le patrimoine génétique des abeilles locales.
Protéger la diversité du miel
Les abeilles du miel (Apis melllifera) se trouvent dans divers écosystèmes, répartis en plusieurs légendes évolutives et au moins 30 sous-espèces. En Europe, il existe une grande partie de cette diversité, avec de nombreuses sous-espèces endémiques divisées en quatre lignées: Afrique (A), Europe centrale et orientale (C), Europe occidentale et septentrionale (M) et Moyen-Orient et Asie centrale (O). Cependant, l'activité humaine a progressivement diminué la diversité génétique des abeilles de miel européennes et leur territoire naturel. Précisément à cause du commerce et de l'importation de reines et de la transhumance à longue distance, une des craintes qui existent est que les populations d'abeilles de miel autochtones adaptées au milieu seront réduites ou perdues, représentées par des abeilles non autochtones ou par simple hybridation. Il est prouvé que les abeilles de miel adaptées au milieu ont une plus grande capacité de survie. Il est donc important de favoriser la croissance des abeilles afin que les abeilles puissent durer longtemps.
En Europe, de nombreux projets ont été mis en place pour la conservation et la croissance des abeilles de miel autochtones, dont les programmes d'amélioration génétique. Le succès dépend du contrôle du champ de fertilisation et du suivi de l'origine génétique de la population. En ce sens, un outil d'analyse génétique rapide, précis et accessible est nécessaire.
Le projet SmartBees a commencé avec l'objectif de développer de nouveaux outils moléculaires permettant de décrire et de conserver la diversité des abeilles à miel européennes. Ainsi, un échantillonnage exhaustif de l'abeille a été conçu un outil composé de marqueurs génomiques à un nucléotide unique qui permet de décrire sa diversité génétique (SNP) et d'identifier la sous-espèce de toute abeille européenne.
Procédure de procédure
22 populations (Figure 1) représentant les quatre lignées évolutionnaires européennes et les 14 sous-espèces qui se trouvent en Europe et dans les régions voisines. Pour l'échantillonnage des populations on a pris 100 ouvrières provenant de ruches non liées entre elles. Au total, plus de 2.000 échantillons, l'échantillonnage le plus complet réalisé à ce jour avec abeille européenne.
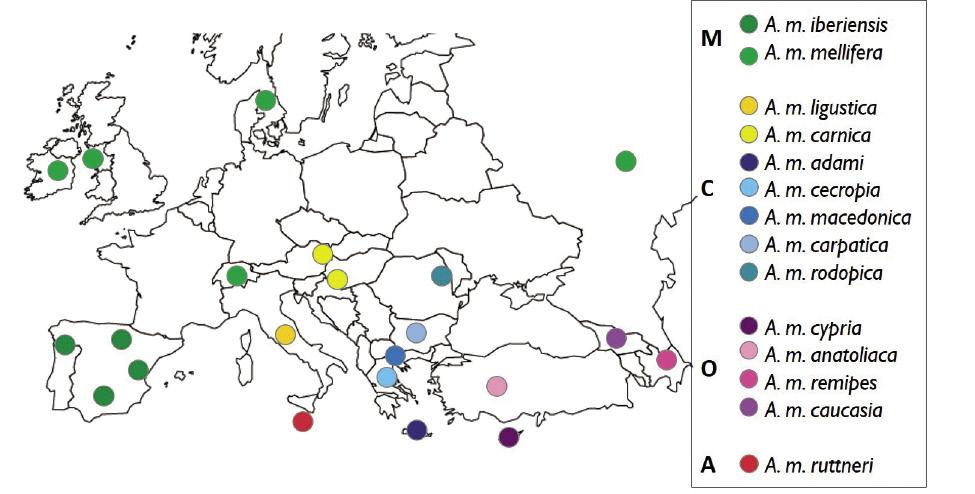
Une fois les échantillons groupés selon la population d'origine, leur ADN a été extrait et séquencé. A partir des données de séquençage, les marqueurs génétiques ont été sélectionnés avec plus d'informations sur l'origine. C'est-à-dire les marqueurs génétiques qui caractérisent le mieux les 14 sous-espèces d'abeilles de miel d'Europe, dépassant au total les 4.000 polymorphismes mononucléotides (SNP). Les résultats de la génotypage ont été représentés en utilisant le diagramme t-SNE. Ce diagramme regroupe les échantillons de la forme la plus compacte possible sur une carte bidimensionnelle, en remplaçant chaque individu par un symbole (figure 2). Avec cette méthode, les abeilles échantillonnées ont été regroupées en groupes isolés (nuages de points) en fonction de leur lignée évolutive ou sous-espèce. La seule sous-espèce montre de la lignée africaine, A. m. ruttneri, se trouve au centre du graphique, parmi les autres groupes. Dans la lignée O, les abeilles A. m. cypria sont apparues complètement séparées de trois abeilles moins différentes entre elles (A. m. anatoliaque, A. m. caucasia et A. m. remipes). Les deux sous-espèces de la lignée M ont été bien différenciées, regroupant les populations de A. m. mellifera en trois sous-groupes. Ces sous-ensembles représentent des régions d'échantillonnage à distance (région de Burzyan, Russie, cluster suprême de A. m. mellife dans la figure 1) ou des régions isolées (île de Læsla, Danemark, cluster inférieur de A. m. mellife). Les échantillons de la lignée C ont été divisés en trois sous-groupes: (i) L'abeille A. m. ligustica, (ii) A. m. carnica avec plusieurs abeilles A. m. carpatica et (iii) un sous-groupe hétérogène d'abeilles A. m. macedonica, A. m. cecropia, A. m. adami, A. m. rodopica et autres A. m. carpatica.
Essais supplémentaires et modèle d'apprentissage automatique
En outre, 1.900 autres abeilles provenant de ruches pour l'élevage de SmartBees ont été génotypées dans toute l'Europe. Avec ces 1.900 échantillons, ainsi que les 2.000 déjà génotypées (plus de 3.900 échantillons au total), ont élaboré un modèle statistique qui classe les abeilles de miel européennes en utilisant des algorithmes d'apprentissage automatique. Ce modèle permet de calculer la probabilité qu'un échantillon soit l'une des 14 sous-espèces européennes. Les modèles d'apprentissage automatiques ont un bon côté, ne sont pas basés sur des hypothèses préalables, donc ils peuvent détecter des différences subtiles. Cette caractéristique a été particulièrement importante pour notre étude, car elle nous a permis de différencier ces grandes quantités de sous-espèces avec une étroite relation génétique. Dans l'article original (Momeni et al. 2021), vous trouverez plus de détails sur les méthodes spécifiques de censure.

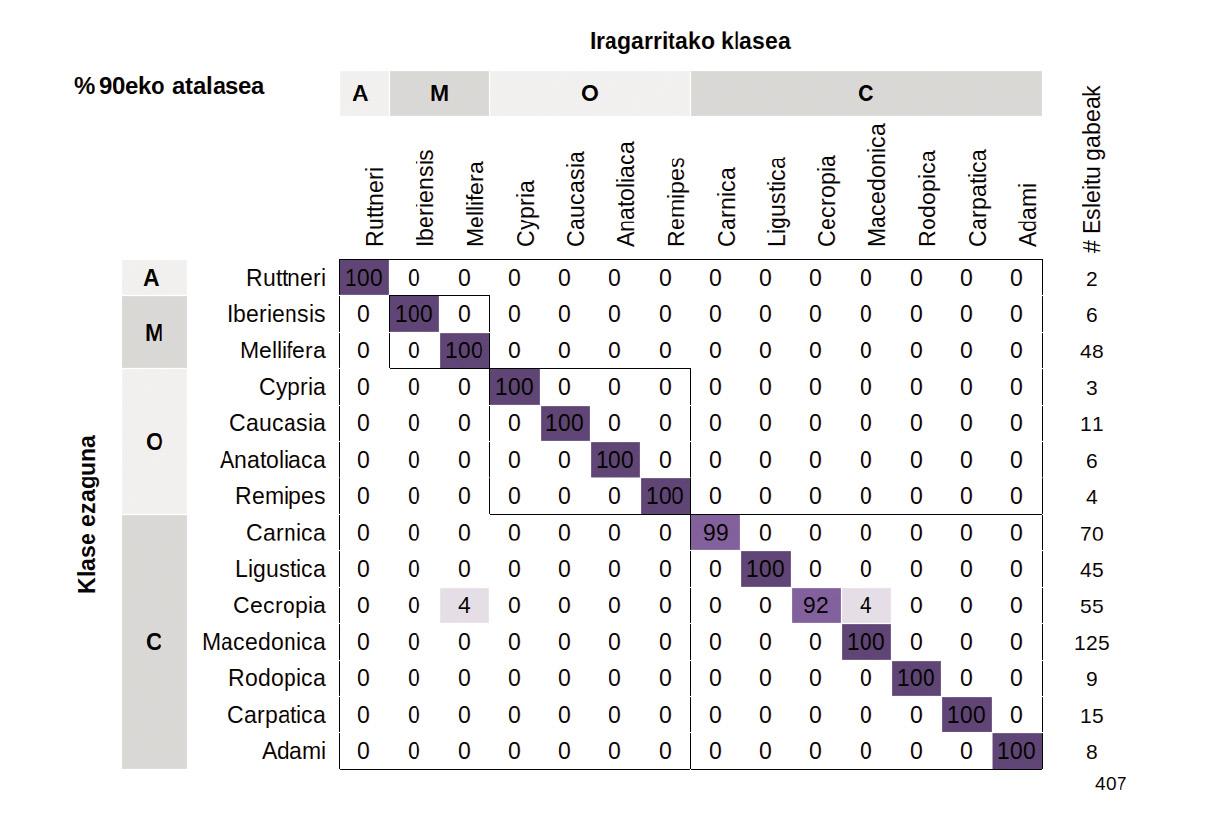
Précision de l'attribution des sous-espèces
Le modèle a correctement alloué la plupart des échantillons avec une précision moyenne de 96,2%. La meilleure façon de visualiser ces résultats est d'utiliser une matrice de confusion, qui représente les pourcentages d'échantillons correctement ou incorrectement classés (Figure 3). Les chiffres de la diagonale centrale indiquent le pourcentage d'échantillons correctement classés, tandis que les triangles supérieur et inférieur représentent les échantillons mal classés. La classification est erronée lorsque les échantillons annoncés par le modèle et étiquetés sont des sous-espèces différentes. Ces erreurs se sont produites, d'une part, lorsque les étiquettes des échantillons testés ont été erronées et, d'autre part, lorsque les différences entre les populations de référence ont été minimes, soit par leur proximité géographique, soit par des interférences humaines.
Pour que ce modèle soit appliqué dans les programmes et programmes de conservation, nous avons établi un seuil minimum de 90%. Ainsi, si la prévision d'un échantillon est inférieure à 90%, elle sera considérée comme « non affectée ». Si elle dépasse la limite, elle sera attribuée à la sous-espèce correspondante.
Défis de la diversité et prochaines étapes
La diversité des abeilles de miel européennes a été un défi majeur pour concevoir un outil de diagnostic de sous-espèces. La divergence des lignées évolutives a rendu possible que la différenciation génétique ait été simple, en utilisant quelques SNP. Cependant, la distinction entre sous-espèces a été plus difficile parce que la divergence entre les espèces du même souverain a été récente et donc génétiquement similaire. En outre, dans certaines régions européennes, les fluctuations du changement de sous-espèces A. mellifera ne sont pas définies, et l'entrée artificielle d'abeilles étrangères a effacé les frontières naturelles entre sous-espèces. Il peut également arriver que les programmes nationaux aient interféré dans le flux naturel des gènes et altéré le patrimoine génétique de la sous-espèce originelle. Par conséquent, certaines sous-espèces ont été facilement classées à l'aide de notre instrument, tandis que d'autres échantillons n'ont pas été affectés. Cependant, il s'agit d'un outil dynamique adaptable qui permet d'améliorer la base de données de référence et/ou d'introduire de nouvelles sous-espèces. En ce sens, compte tenu des recherches en cours, la méthode est applicable à la sicilienne A. m. de la Sicile.
Disponibilité :
Cet instrument permettra de déterminer l'origine génétique de plus d'échantillons, ce qui sera indispensable à bien des égards : les apiculteurs pourront déterminer leur sous-espèce et leur degré d'hybridation des abeilles et donner des avals à leurs produits ; les responsables de la conservation européenne pourront contrôler les taux d'hybridation des ruches en dépôts ; les vétérinaires pourront contrôler le commerce des reines ; et les éleveurs des abeilles assureront leur sous-espèce de reines.
Publication scientifique complète
Remerciements
Nous avons mené cette recherche avec des échantillons d'apiculteurs, d'éleveurs et de collaborateurs. Nous vous remercions de votre collaboration. Le projet SmartBees a été financé par la Commission européenne dans le cadre du programme FP 7 KBBE (02.01.2013, Grant nr. 613960). Melanie Parejo a reçu une subvention du Gouvernement basque (IT1233-19).
Zu idazle
Zientzia aldizkaria

