

El ADN antiguo, nuevo campo de investigación
En este encuentro de dos días se dieron a conocer los descubrimientos de cada grupo y se analizó el futuro de la investigación del antiguo ADN. Entre los participantes se encontraba la profesora e investigadora del campus de Leioa de la UPV, Conchi de la Rúa, con quien hemos podido mantener una conversación. Él nos ha explicado las investigaciones que se han llevado a cabo en un mundo único y su experiencia.
¿Qué es el ADN antiguo?

La mayor parte del material genético de un organismo se almacena en el núcleo de la célula como cromosoma. Los cromosomas están formados por 3.000 millones de nucleótidos que codifican entre 50.000 y 100.000 genes. “Sin embargo, la mayor parte de la cadena de ADN no es un codificador de genes y este carácter neutro hace que el ADN sirva para la investigación genética”, explica Conchi.
Si comparamos el ADN antiguo con el ADN de los seres vivos, hay algunas diferencias notables. En primer lugar, la cantidad de ADN antiguo que se obtiene en las extracciones es muy baja respecto a la que se puede obtener de un vivo, entre 0,1 y 1%. Y en segundo lugar, la cadena de ADN resultante está fragmentada en cadenas de 100 pares de bases, mientras que en seres vivos se pueden obtener cadenas de miles de pares de bases.
Estas diferencias se deben a los procesos de degradación que se producen en las muestras. “Los efectos de oxidación e hidrólisis que se producen tras la muerte del organismo provocan la ruptura de la cadena de ADN, siendo de gran importancia la conservación del organismo para la calidad de las muestras en un entorno seco y sin oxígeno”, explica. Por este motivo, la edad límite del ADN antiguo se sitúa en 100.000 años.
ADN mitocondrial
Debido al escaso número de antiguas moléculas de ADN recuperadas, la mayoría de los investigadores se han dirigido a trabajar con ADN mitocondrial en el caso del antiguo ADN. Como el ADN nuclear es único en cada célula, el ADN mitocondrial es abundante. Esta abundancia aumenta la supervivencia y las posibilidades de recuperación del ADN.
En el caso de la mitocondria, la longitud de la cadena de ADN es de 16.500 pares de bases nitrogenadas, lo que supone el 0,0005% del genoma humano. El ADN de las mitocondrias humanas y de varios vertebrados está secuenciado y en esta secuencia se encuentra una zona denominada área de control. Esta zona equivalente en base 1.100 es neutra, de manera que las mutaciones sólo aparecen bajo la influencia del tiempo y no por la selección natural. Además, el punto de control soporta el 90% de las mutaciones de ADN mitocondrial.

A estas particularidades hay que añadir otra: El ADN mitocondrial sólo se recibe de su madre. “El ADN mitocondrial se encuentra en la cola de los espermatozoides, y durante la fecundación del óvulo la cola queda fuera –dice Conchi–, quedando así fuera el ADN mitocondrial masculino”.
Esto supone reducir a la mitad el área de estudio y facilitar el seguimiento de la marca de deriva genética.
Iniciación
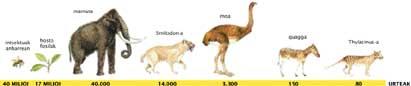
La primera investigación del ADN antiguo la realizaron en 1984 con un animal que desapareció hace 150 años. Una antigua secuencia de ADN se obtuvo por primera vez del tejido muscular seco de un equus quagga. De esta secuencia, demostraron la relación evolutiva entre el quagga y los parientes más cercanos de hoy, la cebra.
Un año después, en 1985, otros investigadores recuperaron el ADN de las células superficiales de las momias. De las 110 momias seleccionadas, 23 estaban mejor conservadas y sólo dos de ellas obtuvieron ADN. Este paso fue el inicio del estudio del antiguo ADN humano.
Continuando con las investigaciones, a mediados de la década de los 90 se consiguió secuenciar fragmentos de ADN de vertebrados ya desaparecidos, como el Thylacinus o el lobo martsupial (120 años), el Smilodón o tigre ‘con dientes de sable’ (14.000 años), el mamut oscuro (10.000-50.000 años) y el model (3.300 años).
Además, se estudió una hoja de magnolia llamada Ma (17 millones de años) y varios insectos almacenados en la ámbar, pero sus estudios no se consideran actualmente fiables. “No se ha conseguido repetir los resultados obtenidos en estos laboratorios ni en el mismo ni en otros, lo que invalida los resultados”, afirma Conchi. “Sin embargo, quedó demostrado que el ADN antiguo era recuperable”.
En 1986 se da un paso más importante al lanzar el nuevo método de análisis PCR. Esta técnica revolucionó la investigación del ADN y permitió el descubrimiento de los genes mitocondriales.
Método PCR
La reacción en cadena o método PCR de la polimerasa consiste en sintetizar millones de copias de un pequeño fragmento de ADN. En este proceso cíclico
Parte de la cadena de ADN se duplica exponencialmente hasta disponer de muestras suficientes para aplicar los diferentes métodos de análisis. El PCR requiere el diseño de cadenas de ADN cortas con función limitante, complementarias de la cadena de ADN objeto de estudio. La función de estos limitadores es fijar la longitud de la cadena de ADN a duplicar, ya que se añaden al principio y al final. A continuación, la enzima denominada Taq polimerasa realiza el doblaje de la cadena de ADN.
Así, el método PCR permitió ampliar los estudios a huesos y dientes y, al ser un material corriente procedente de excavaciones arqueológicas, se abrió la puerta al estudio de la historia biológica de los siglos.
Problemas metodológicos y contaminación
Sin embargo, la capacidad de duplicación del método PCR se convierte en un problema en el caso del antiguo ADN. El ADN antiguo tiene un alto riesgo de contagio al entrar en contacto con cualquier elemento externo. Teniendo en cuenta esto, y dado que las antiguas cadenas de ADN que se recuperan son cortas, el método de PCR tiene una gran probabilidad de copiar fragmentos de ADN externos. Por ello, es imprescindible adoptar una serie de medidas para asegurar el resultado del doblaje.
En primer lugar, se debe asegurar el aislamiento físico del laboratorio para que no afecte a elementos externos; en segundo lugar, se debe utilizar únicamente el equipamiento destinado a la recuperación del ADN antiguo y, finalmente, se debe asegurar la total esterilidad de las muestras a utilizar. Estas muestras se suelen extraer de los granos que mejor se conservan, generalmente usando huesos y dientes. “La cantidad de ADN antiguo disponible extraído de la pulpa es mayor que la que se obtiene del resto de tejidos o huesos, y además en los yacimientos suele ser una pieza común”.
Además de todas las medidas mencionadas, los estudios deben repetirse tanto en el mismo laboratorio como en diferentes laboratorios, utilizando más de una muestra del mismo ejemplar, lo que asegura la existencia o no de contaminación. Finalmente, para homologar la antigua secuencia de ADN, se debe comparar con el universo de secuencias conocidas que se almacenan en las bases de datos, analizando posteriormente los resultados en un contexto filogenético adecuado y comparándolos con las secuencias de otras especies vivas o muertas de un mismo grupo taxonómico.
¿Qué métodos utilizáis para los exámenes?
“Se pueden utilizar dos métodos, uno de secuenciación y otro de análisis de enzimas restrictivas”.

La secuenciación es conocer la localización de las bases nitrogenadas responsables de la información genética en la cadena de ADN. Este método automático con varios pasos consiste en una amplificación especial de la cadena de ADN, donde se obtienen fragmentos fluorescentes de diferentes tamaños. A continuación se distribuyen mediante electroforesis, es decir, se separan entre sí en un gel sobre el que se aplica un campo eléctrico. La velocidad de migración depende del tamaño. En este proceso un lector láser detecta la emisión fluorescente de cada parte, que depende de la última base nitrogenada que se añadió en el proceso de PCR. En consecuencia, se conoce la base nitrogenada en función de la fluorescencia que se desprenda.
Con las enzimas restrictivas se analizan las secuencias de destinos. “Las secuencias de meta son cadenas cortas de par nitrogenado en la cadena ADN”. Las enzimas restrictivas son capaces de romper la cadena de ADN desde donde se encuentran las secuencias de destino. Estas secuencias de meta, que tienen una longitud de 4-6 pares de bases nitrogenadas (pb), son diferentes para cada enzima reductora, es decir, una enzima reductora debe encontrar una secuencia de metas para poder romper la cadena de ADN, que en muchas ocasiones no existe. “Viendo si la cadena ADN reacciona o no con diferentes enzimas, podemos saber si tiene diferentes secuencias de destino y si se trata o no de secuencias de destino, según los diferentes patrones existentes, podemos ver genéticos relacionales”. Metodológicamente esta segunda técnica es más sencilla y rápida, lo que permite obtener más resultados positivos.
Futuro próximo
En los últimos años la investigación del ADN antiguo se ha centrado en la búsqueda de relaciones filogenéticas, en el estudio de la variabilidad genética a lo largo del tiempo o en la investigación de cambios demográficos. Todo ello se debe a los avances en biología molecular y al desarrollo de la técnica PCR. Sin embargo, todavía quedan obstáculos como el riesgo de contaminación y la mala conservación del ADN antiguo.
De cara al futuro, el estudio está orientado a mejorar la metodología de extracción del ADN y a aumentar el número de muestras antiguas. Además, las nuevas investigaciones se centrarán en profundizar en la investigación del ADN nuclear. Estos indicadores permitirán no sólo analizar la información recibida de su madre, sino también la de su padre. ¿El futuro nos dará el límite de esta ciencia?
I España del ADN antiguo. CongresoEn la Residencia de la Vidriera de la Universidad Autónoma de Madrid, situada en Miraflores de la Sierra, se ha celebrado la I Edición Española del ADN antiguo. Congreso. Entre los invitados se encontraban expertos de 15 centros de investigación dedicados a la biología evolutiva, paleobiología, antropología, genética molecular, forense e ictología. Estos grupos de investigadores se plantearon conocer y profundizar en los objetivos comunes de su ciencia. Entre estos objetivos se analizó la creación de un grupo de trabajo sobre el ADN antiguo. Este grupo se encargaría de establecer un procedimiento básico para garantizar la calidad de las investigaciones en esta ciencia. Otro de los objetivos era reforzar las relaciones entre laboratorios, impulsando la colaboración entre los grupos investigadores. Por último, se comentó la intención de dar a conocer las potenciales aplicaciones de este campo científico para que la gente tenga en cuenta las posibles aplicaciones de la investigación del ADN antiguo en otros campos. |
¿Qué hay en Euskal Herria?
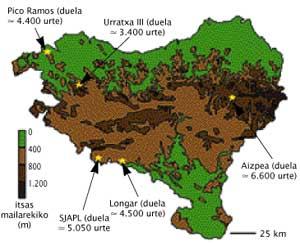
Conchi de la Rúa y su equipo dirigen sus investigaciones a la antropología. El método de las enzimas restrictivas ha permitido clasificar el 97% de las muestras libres de contaminación de diversos yacimientos vascos. Aunque el porcentaje parece alto, hay que decir que tan sólo el 25% de las muestras recuperadas están representadas, ya que sólo se han analizado los dientes en perfecto estado para evitar el riesgo de contaminación.
Entre los descubrimientos se han obtenido datos para oponerse a las teorías de algunos investigadores italianos. Estos investigadores italianos informaban sobre un grupo que hace entre 10.000 y 15.000 años se extendió desde el País Vasco a Europa. El objetivo de esta teoría era explicar la abundancia de un grupo genético existente actualmente en la sociedad vasca, el denominado V Haplotalde. Los trabajos de los investigadores vascos han demostrado que existe la ausencia de este grupo en las muestras de ADN antiguas, es decir, que no hay indicios de esa época del haplotalde. ¿Qué significa esto? “Que este grupo de Haplots no se creó aquí porque si fuera de aquí debería aparecer en muestras con menos de 10.000 años de antigüedad”.
Conchi y sus compañeros ponen el punto de vista en la deriva genética para explicar la abundancia de este grupo. “La mutación que define el Haplotaldea pudo alcanzar una tasa elevada en un grupo de población, como es el caso de Gipuzkoa, en una época en la que la población era reducida, y los crecimientos de población posteriores permitieron ampliar dicha mutación”.
Zu idazle
Zientzia aldizkaria

